Decoding LSER Solute Descriptors: A Comprehensive Guide to Vx, E, S, A, B, L for Pharmaceutical Research
This article provides a comprehensive exploration of Linear Solvation Energy Relationship (LSER) solute descriptors—Vx, E, S, A, B, L—tailored for researchers and professionals in drug development.
Decoding LSER Solute Descriptors: A Comprehensive Guide to Vx, E, S, A, B, L for Pharmaceutical Research
Abstract
This article provides a comprehensive exploration of Linear Solvation Energy Relationship (LSER) solute descriptors—Vx, E, S, A, B, L—tailored for researchers and professionals in drug development. It covers the fundamental chemical significance of each parameter, methodological approaches for their determination, troubleshooting common challenges in descriptor application, and validation techniques against experimental data. By integrating theoretical foundations with practical applications, this guide serves as an essential resource for leveraging LSERs in predicting solute partitioning, solubility, and membrane permeability to optimize pharmaceutical compounds.
Understanding LSER Solute Descriptors: A Deep Dive into Vx, E, S, A, B, L Fundamentals
Linear Solvation Energy Relationships (LSERs) represent a cornerstone quantitative model in physical organic and pharmaceutical chemistry for predicting the partitioning behavior of solutes in different chemical environments. The model provides a powerful framework for understanding and predicting how molecules distribute themselves between phases, a process fundamental to drug absorption, distribution, and clearance. The Abraham solvation parameter model, as LSER is formally known, has demonstrated remarkable success as a predictive tool across chemical, biomedical, and environmental applications [1]. Its robustness stems from its ability to deconstruct complex solvation phenomena into contributions from fundamental, chemically intuitive molecular interactions.
The theoretical foundation of LSER rests on Linear Free Energy Relationships (LFER), which posit that free-energy-related properties of a solute can be correlated with molecular descriptors representing its capacity for specific interaction types [1]. In thermodynamic terms, the partitioning of a solute between two solvents is equivalent to the difference in two gas/liquid solution processes [2]. This process is modeled as the sum of an endoergic cavity formation/solvent reorganization process and exoergic solute-solvent attractive forces [2]. The remarkable linearity observed in these relationships, even for strong specific interactions like hydrogen bonding, has been verified through the combination of equation-of-state solvation thermodynamics with the statistical thermodynamics of hydrogen bonding [1].
The Abraham LSER Model and Solute Descriptors
The most widely accepted symbolic representation of the LSER model, as proposed by Abraham, is given by the equation:
SP = c + eE + sS + aA + bB + vV [2]
In this equation, SP represents any free energy related property. In pharmaceutical contexts, this is most often the logarithm of a partition coefficient (log P) or retention factor (log k') [2]. The lowercase letters (e, s, a, b, v) are system coefficients reflecting the complementary properties of the phases between which partitioning occurs, while the uppercase letters (E, S, A, B, V) are solute descriptors that quantify the solute's ability to participate in specific intermolecular interactions [2]. An alternative form of the equation uses L instead of V for gas-to-solvent partitioning: log (KS) = ck + ekE + skS + akA + bkB + lkL [1].
The following table provides a detailed explanation of each solute descriptor and its molecular interpretation:
Table 1: Abraham LSER Solute Descriptors and Their Molecular Significance
| Descriptor | Name | Molecular Interpretation | Role in Solvation |
|---|---|---|---|
| E | Excess molar refraction | Electron pair interactions and polarizability [3] | Measures the solute's ability to interact through π- and n-electrons [2] |
| S | Solute dipolarity/polarizability | Dipolarity and polarizability [3] | Characterizes the solute's ability to engage in dipole-dipole and dipole-induced dipole interactions [2] |
| A | Hydrogen bond acidity | Overall hydrogen-bond donating ability [3] | Quantifies the solute's effectiveness as a hydrogen bond donor [2] |
| B | Hydrogen bond basicity | Overall hydrogen-bond accepting ability [3] | Quantifies the solute's effectiveness as a hydrogen bond acceptor [2] |
| V | McGowan characteristic volume | Molecular size [3] | Represents the endoergic cost of forming a cavity in the solvent [2] |
| L | Gas-liquid partition coefficient | Partition coefficient in n-hexadecane at 298 K [1] | Alternative to V for gas-to-solvent partitioning; related to dispersion interactions [1] |
These descriptors can be obtained experimentally through various physicochemical measurements, including gas-liquid chromatographic data and water-solvent partition coefficients [3]. Increasingly, computational approaches are also available for their estimation from molecular structure alone [4].
LSERs in Pharmaceutical Applications
Predicting Drug Partitioning and Absorption
In pharmaceutical development, LSERs provide invaluable predictive capabilities for key absorption, distribution, metabolism, and excretion (ADME) parameters. The model has been successfully applied to predict critical properties including aqueous solubility (log S₍w₎), various water-solvent partition coefficients (log P₍s₎), and air-solvent partition coefficients (log L₍s₎) [4]. These properties directly influence a drug's membrane permeability, bioavailability, and distribution patterns within the body. The ability to predict such properties from molecular structure alone during early development stages enables more efficient lead optimization and candidate selection.
Leachables and Extractables Assessment
LSERs have emerged as particularly valuable for addressing pharmaceutical packaging challenges, specifically in predicting the partitioning of potential leachables between plastic materials and pharmaceutical solutions. This application is critical for ensuring drug product safety and regulatory compliance. A recently developed LSER model for partitioning between low-density polyethylene (LDPE) and water demonstrates the precision achievable:
logK₍i,LDPE/W₎ = −0.529 + 1.098E − 1.557S − 2.991A − 4.617B + 3.886V [5]
This model, calibrated using 159 chemically diverse compounds, exhibited exceptional accuracy (n = 156, R² = 0.991, RMSE = 0.264) and outperformed traditional log-linear models, particularly for polar compounds with significant hydrogen-bonding propensity [5]. The model enables identification of maximum (worst-case) levels of leaching in support of chemical safety risk assessments [5].
Chromatographic Method Development
In pharmaceutical analysis, LSERs extensively characterize retention mechanisms in high-performance liquid chromatography (HPLC). The coefficients derived from LSER analysis reveal how specific stationary phases interact with different solute functionalities, guiding the rational selection of chromatographic conditions for method development [3]. Studies have demonstrated that the most important parameters influencing retention are typically the solute volume (V) and hydrogen bond acceptor basicity (B) [3]. This understanding allows chromatographers to optimize separations based on the specific molecular properties of analytes rather than through purely empirical approaches.
Experimental Protocols and Methodologies
Determining Solute Descriptors
Experimental determination of LSER descriptors requires careful measurement of partition coefficients in well-characterized systems. The following protocol outlines the standard approach for determining descriptors for new chemical entities:
Measure gas-chromatographic retention indices on at least 6-8 stationary phases of different polarities to obtain the L descriptor [4].
Determine water-solvent partition coefficients (log P) for a minimum of 6-8 solvent systems with known LSER coefficients [4]. The shake-flask method is commonly employed, though the microshake-flask method has been introduced for compounds with limited availability [4].
Validate descriptor consistency by comparing predicted and measured partition coefficients in additional solvent systems not used in the initial regression [2].
For compounds with limited solubility, consider alternative approaches including reversed-phase HPLC retention times and calculated descriptor values from molecular structure using validated software tools [4].
Developing System-Specific LSER Models
When creating new LSER models for specific pharmaceutical applications, the following methodological considerations are essential:
Select a chemically diverse training set of solutes spanning a wide range of molecular weights, hydrophobicity, and hydrogen-bonding capabilities [5]. The training set should be representative of the "chemical space" of compounds relevant to the application domain [5].
Ensure experimental data quality through appropriate replication, calibration, and validation of analytical methods. For partition coefficient measurements, equilibrium must be verified and mass balance confirmed [5].
Perform multiple linear regression analysis using the Abraham equation to determine system coefficients. Statistical evaluation should include R², root mean square error (RMSE), and leave-one-out cross-validation [2].
Validate the model using an independent test set not included in the initial regression. For the LDPE/water partitioning model, validation with 52 compounds (approximately 33% of the total observations) yielded R² = 0.985 and RMSE = 0.352 [6].
Figure 1: LSER Model Development Workflow for Pharmaceutical Applications
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful application of LSERs in pharmaceutical research requires access to well-characterized materials and computational resources. The following table outlines essential components of the LSER toolkit:
Table 2: Essential Research Tools for LSER Studies in Pharmaceutical Science
| Tool/Reagent | Function/Purpose | Pharmaceutical Relevance |
|---|---|---|
| Reference Solutes | Chemically diverse compounds with known descriptors for system calibration [2] | Enables characterization of new partitioning systems and stationary phases |
| Well-Characterized Solvents | Solvents with known LSER system coefficients for descriptor determination [4] | Provides standardized environments for measuring solute-specific interactions |
| Chromatographic Columns | Stationary phases with known LSER characteristics (e.g., C18, alkylamide, cholesterol) [3] | Facilitates determination of solute descriptors via HPLC retention measurements |
| Abraham Descriptor Database | Curated database of solute descriptors for known compounds [6] | Provides essential parameters for predictive modeling without experimental work |
| QSPR Prediction Software | Computational tools for predicting LSER descriptors from molecular structure [4] | Enables descriptor estimation for novel compounds when experimental data is lacking |
| Polymer Materials | Characterized polymeric phases (LDPE, PDMS, POM) with known LSER models [6] | Allows prediction of leachables partitioning from packaging materials into formulations |
Current Research and Future Perspectives
Recent advances in LSER applications continue to expand their utility in pharmaceutical sciences. The development of Partial Solvation Parameters (PSP) based on equation-of-state thermodynamics represents a promising approach for extracting thermodynamic information from LSER databases [1]. These parameters are designed to facilitate the exchange of information between QSPR-type databases and equation-of-state developments, potentially enabling estimation of solvation properties over broad ranges of temperature and pressure [1].
Comparative studies of polymer sorption behavior using LSER system parameters have revealed important differences between materials. While low-density polyethylene (LDPE) exhibits predominantly hydrophobic character, polymers like polyoxymethylene (POM) with heteroatomic building blocks demonstrate stronger sorption for polar, non-hydrophobic compounds [6]. Such insights guide the selection of appropriate packaging materials for specific drug formulations.
Ongoing research addresses the challenge of predicting LSER descriptors for novel chemical entities. The availability of "rule of thumb" estimation methods for variable values based on molecular functional groups has improved accessibility of the LSER approach [7]. Furthermore, the introduction of user-friendly software tools for descriptor calculation and the creation of freely accessible, curated databases promise to broaden LSER adoption in pharmaceutical development [4] [6].
As pharmaceutical research increasingly embraces in silico approaches, LSERs provide a thermodynamically grounded, chemically intuitive framework for predicting critical physicochemical properties. Their ability to decomplexify solvation phenomena into fundamental interactions makes them uniquely valuable for rational drug design and formulation optimization.
Figure 2: LSER Model Components and Their Relationships in Pharmaceutical Prediction
Linear Solvation Energy Relationships (LSERs) represent a cornerstone of quantitative structure-property relationship (QSPR) modeling, providing a powerful predictive framework for understanding solvation phenomena across chemical, biological, and environmental sciences. The widely accepted Abraham model formulation offers a sophisticated mathematical framework that quantifies how solute properties interact with their solvation environment, enabling researchers to predict partition coefficients, solubility, retention in chromatography, and other free-energy-related properties with remarkable accuracy [2]. This model's robustness stems from its foundation in linear free-energy relationships (LFER), which establish quantitative connections between molecular structure and thermodynamic behavior.
The LSER approach operates on the fundamental principle that solvation processes can be deconstructed into discrete, chemically meaningful interactions, each contributing additively to the overall free energy change [1]. This conceptual framework allows researchers to move beyond empirical correlations toward mechanistically interpretable models. The LSER model and its associated database are exceptionally rich in thermodynamic information about intermolecular interactions, which, when properly extracted, provides invaluable insights for various thermodynamic developments and applications [1]. This technical guide deconstructs the theoretical foundations, solute descriptors, and experimental methodologies underpinning LSERs, with particular emphasis on their significance for researchers in pharmaceutical development and analytical chemistry.
The LSER Equation: Fundamental Principles and Thermodynamic Basis
Mathematical Formulation of the Abraham Model
The most current and widely adopted symbolic representation of the LSER model, as proposed by Abraham, is expressed by the following equation:
SP = c + eE + sS + aA + bB + vV
In this foundational equation, SP represents any free-energy-related property, which in chromatographic applications most commonly corresponds to log k' (where k' is the retention factor) [2]. The lowercase coefficients (e, s, a, b, v) and constant (c) are system-specific parameters determined through multiparameter linear least squares regression analysis of datasets comprising solutes with known descriptor values [2]. These coefficients quantify the solvent's complementary responsiveness to each type of solute interaction.
The model's thermodynamic foundation lies in its interpretation of the solvation process. The partitioning of a solute between two solvents is thermodynamically equivalent to the difference in two gas/liquid solution processes [2]. For gas-liquid partitioning, the process is conceptually modeled as the sum of an endoergic cavity formation/solvent reorganization process and exoergic solute-solvent attractive forces [2]. This physical interpretation provides a chemically intuitive framework for understanding the contributions of the various terms in the LSER equation.
Thermodynamic Basis of Linearity
A fundamental question surrounding LSER models concerns the thermodynamic basis for the observed linearity, particularly for strong specific interactions like hydrogen bonding. Recent research combining equation-of-state solvation thermodynamics with the statistical thermodynamics of hydrogen bonding has verified that there is indeed a sound thermodynamic basis for LFER linearity [1]. This insight validates the model's application even for complex interaction types and provides deeper understanding of the thermodynamic character and content of the coefficients and terms in the LSER equations.
The LSER formalism has been successfully extended beyond free energy predictions to encompass other thermodynamic properties. Enthalpies of solvation, for instance, can be handled through a parallel linear relationship of the form [1]:
ΔHS = cH + eHE + sHS + aHA + bHB + lHL
This extension demonstrates the versatility of the LSER approach and enables researchers to extract valuable thermodynamic information about intermolecular interactions for solute/solvent systems where both LSER descriptors and LFER coefficients are available [1].
Deconstructing the LSER Solute Descriptors
Comprehensive Descriptor Definitions and Chemical Significance
The capital letters in the LSER equation (E, S, A, B, V) represent solute-specific molecular descriptors that quantify distinct aspects of a molecule's interaction potential. A deep understanding of these parameters' physico-chemical basis is essential for proper application and interpretation of LSER models [2].
Table 1: LSER Solute Descriptors and Their Chemical Significance
| Descriptor | Full Name | Chemical Interpretation | Measurement Basis | Molecular Feature Quantified |
|---|---|---|---|---|
| V | McGowan's Characteristic Volume | Molecular size influencing cavity formation | Computational from molecular structure | Energy cost of displacing solvent to create cavity |
| E | Excess Molar Refraction | Polarizability from π- and n-electrons | Measured refraction compared to alkane analog | Dispersion interaction capability |
| S | Dipolarity/Polarizability | Dipolarity with polarizability contribution | Solvatochromic comparison method | Strength of dipole-dipole and dipole-induced dipole interactions |
| A | Hydrogen Bond Acidity | Hydrogen bond donating ability | Solvatochromic measurement or computational | Strength as hydrogen bond donor |
| B | Hydrogen Bond Basicity | Hydrogen bond accepting ability | Solvatochromic measurement or computational | Strength as hydrogen bond acceptor |
| L | Gas-Hexadecane Partition Coefficient | Overall lipophilicity at molecular level | Experimental partition coefficient in n-hexadecane at 298 K | General dispersion interactions |
The development and physico-chemical basis of these solute parameters establishes their fundamental meaning and proper application [2]. Each descriptor corresponds to a specific type of solute-solvent interaction that contributes to the overall solvation energy, allowing for a nuanced understanding of molecular recognition processes in solution.
System Coefficients and Their Interpretation
The lowercase coefficients in the LSER equation (e, s, a, b, v) are system-specific parameters that reflect the solvent's responsiveness to each type of solute interaction. These coefficients are considered to correspond to the complementary effect of the phase (solvent) on solute-solvent interactions and contain chemical information on the solvent/phase in question [1]. In practical applications, these coefficients are determined via multiple linear regression analysis of experimental data for a diverse set of solutes with known descriptor values [2].
For partition processes between two condensed phases, the LSER relationship takes the form [1]:
log (P) = cp + epE + spS + apA + bpB + vpVx
Where P represents the partition coefficient between two solvents (e.g., water-to-organic solvent or alkane-to-polar organic solvent). For gas-to-solvent partitioning, the relationship becomes [1]:
log (KS) = ck + ekE + skS + akA + bkB + lkL
The remarkable feature of these equations is that the coefficients are solvent (phase or system) descriptors and are not influenced by the solute [1]. This characteristic enables the prediction of solute behavior in known systems and facilitates the rational selection of solvent systems for specific separation needs in pharmaceutical development.
Experimental Protocols for LSER Parameter Determination
Solvatochromic Method for Solute Parameter Measurement
The determination of solute descriptors relies on a combination of experimental measurements and computational approaches. For the S (dipolarity/polarizability), A (hydrogen bond acidity), and B (hydrogen bond basicity) parameters, solvatochromic methods provide a robust experimental approach [8].
Protocol: Solvatochromic Measurement of Kamlet-Taft Parameters
Sample Preparation: Prepare solutions of solvatochromic indicator probes (e.g., 4-nitroaniline) in the solvent systems of interest at controlled concentrations (approximately 10 µM). Use volatile solvents for stock solutions that can be evaporated and replaced with the test solvent [8].
Spectrophotometric Analysis: Record UV-Vis absorption spectra over an appropriate wavelength range (e.g., 300-700 nm) using a precision spectrophotometer. Maintain constant temperature (typically 298.15 K) using thermostated cell holders [8].
Data Processing: Calculate the molar electronic transition energy, ET, from the maximum absorption wavelength using the relationship: ET(kcal·mol⁻¹) = 2.85915 × νmax(cm⁻¹) where νmax is the wavenumber of maximum absorption [8].
Multiple Linear Regression: Correlate the ET values with solvent parameters using the LSER equation: ET = A₀ + aa + bβ + pπ where a, β, and π are the Kamlet-Taft solvatochromic parameters representing hydrogen-bond donor acidity, hydrogen-bond acceptor basicity, and dipolarity/polarizability, respectively [8].
Statistical Validation: Evaluate the regression model using statistical parameters including squared correlation coefficient (r²), F-statistic values, and standard deviations of regression coefficients to identify the optimal model [8].
This protocol enables the determination of solvent parameters that can be correlated with Abraham parameters, establishing a bridge between different LSER formalisms.
Chromatographic Method for System Characterization
Chromatographic techniques provide a powerful approach for characterizing system parameters (coefficients) in the LSER equation.
Protocol: Determination of LSER System Coefficients by Chromatography
Column Selection and Conditioning: Select the chromatographic stationary phase of interest and condition according to manufacturer specifications. For reversed-phase systems, ensure equilibration with mobile phase.
Test Solute Selection: Curate a diverse set of test solutes (30-40 compounds) spanning a wide range of descriptor values to ensure a well-conditioned regression. Include compounds with varied hydrogen bonding capabilities, polarizabilities, and molecular sizes [2].
Chromatographic Measurement: Determine retention factors (k') for each test solute under isocratic conditions. Ensure sufficient replication to establish measurement precision (typically RSD < 2%).
Descriptor Assignment: Obtain solute descriptors (E, S, A, B, V) from authoritative databases or through experimental determination for each test compound.
Multiple Linear Regression: Perform multiparameter linear regression of log k' against the solute descriptors to obtain the system-specific coefficients (e, s, a, b, v) and constant (c).
Model Validation: Validate the model using leave-one-out cross-validation or external test sets to ensure predictive capability. The model should cover at least 80% of the variance in the training and test sets for reliable application [9].
This methodology allows for the characterization of chromatographic systems, enabling rational method development in pharmaceutical analysis and quality control.
Figure 1: Experimental Workflow for LSER Parameter Determination. This diagram illustrates the parallel pathways for determining solute descriptors (solvatochromic method) and system coefficients (chromatographic method).
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of LSER research requires specific reagents, materials, and instrumentation. The following table details essential components of the LSER research toolkit.
Table 2: Essential Research Reagents and Materials for LSER Studies
| Category | Specific Items | Function/Application | Technical Specifications |
|---|---|---|---|
| Solvatochromic Indicators | 4-Nitroaniline, Reichardt's dye, Nitroanisoles | Probe for specific solvent interactions; determination of S, A, B parameters | High purity (>99%); wavelength calibration standards |
| Chromatographic Test Solutes | Alkylbenzenes, nitroalkanes, anilines, phenols, polycyclic aromatic hydrocarbons | Characterization of system coefficients; must span descriptor space | 30-40 diverse compounds; known descriptor values |
| Reference Solvents | n-Hexadecane, water, alkanols, ethers, dimethyl sulfoxide | Reference systems for partition coefficient measurements; calibration standards | HPLC grade; controlled humidity for aqueous systems |
| Chromatographic Equipment | HPLC system with UV-Vis detector, analytical columns | Determination of retention factors for system characterization | Precision: RSD < 2% for retention times |
| Spectroscopic Equipment | UV-Vis spectrophotometer, quartz cuvettes, temperature controller | Solvatochromic measurements; determination of transition energies | Wavelength accuracy: ±0.5 nm; temperature control: ±0.1°C |
| Computational Resources | LSER database software, statistical analysis package, molecular modeling suite | Regression analysis; descriptor calculation; model validation | Multiple linear regression with cross-validation capability |
This toolkit enables researchers to implement the experimental protocols described previously and generate high-quality LSER data for both fundamental studies and applied pharmaceutical research.
Advanced Applications and Recent Methodological Developments
Integration with Equation-of-State Thermodynamics
Recent advances in LSER methodology have focused on integrating the approach with equation-of-state thermodynamics to extract more detailed thermodynamic information. The development of Partial Solvation Parameters (PSP) represents a significant innovation designed to facilitate the extraction of thermodynamic information from LSER databases [1]. PSPs are characterized by their equation-of-state thermodynamic basis, which permits estimation over a broad range of external conditions [1].
This integration enables the estimation of key thermodynamic quantities beyond free energy, including the enthalpy change (ΔH) and entropy change (ΔS) upon formation of hydrogen bonds [1]. The hydrogen-bonding PSPs (σa and σb) reflect the acidity and basicity characteristics of molecules, respectively, while the dispersion PSP (σd) reflects weak dispersive interactions, and the polar PSP (σp) collectively reflects remaining Keesom-type and Debye-type polar interactions [1].
Artificial Intelligence and Machine Learning Enhancements
The field of LSER modeling is being transformed by artificial intelligence and machine learning approaches, which offer powerful alternatives to traditional regression methods. Recent developments include dataset-based machine learning and hybrid quantum mechanical/machine learning models that achieve superior accuracy in free energy predictions with reduced computational costs [10].
Machine learning models now enable rapid pKa predictions across a wide range of diverse solvents, and the integration of thermodynamic principles into machine learning frameworks allows for accurate and consistent macro-micro pKa prediction [10]. Graph-convolutional neural networks demonstrate high accuracy in reaction outcome prediction with interpretable mechanisms, representing a significant advancement over traditional LSER approaches for complex systems [10].
These AI/ML enhancements maintain the chemical interpretability of traditional LSER approaches while significantly expanding their predictive capability and application domain, particularly in pharmaceutical research where prediction of solvation-related properties is crucial for drug design and development.
The Linear Solvation Energy Relationship equation represents a sophisticated framework that deconstructs solvation phenomena into chemically meaningful interaction terms. The Abraham model, with its well-defined solute descriptors (V, E, S, A, B, L) and system-specific coefficients, provides both predictive power and mechanistic insight into molecular interactions in solution. The experimental protocols for parameter determination, particularly solvatochromic and chromatographic methods, provide robust approaches for characterizing both solute properties and system responses.
Recent advances integrating LSER with equation-of-state thermodynamics and machine learning approaches are expanding the methodology's capabilities, enabling more detailed thermodynamic analysis and enhanced predictive accuracy. For researchers in drug development, these developments offer increasingly powerful tools for predicting solubility, permeability, and other pharmaceutically relevant properties, ultimately supporting more efficient drug design and development processes. The continued evolution of LSER methodology promises to further bridge the gap between empirical correlation and fundamental molecular understanding in solvation science.
The Solvation Parameter Model, often expressed through Linear Solvation Energy Relationships (LSER), is a powerful quantitative structure-property relationship (QSPR) tool for predicting a wide range of chemical, biological, and environmental processes [11]. The model characterizes intermolecular interactions using a set of solute descriptors. The most current and widely accepted form of the Abraham model is represented by the equation [2] [12] [1]:
SP = c + eE + sS + aA + bB + vV
In this equation, SP is a free-energy related property, such as the logarithm of a partition coefficient or retention factor in chromatography [2]. The capital letters represent the solute descriptors:
- E: Excess molar refraction
- S: Dipolarity/Polarizability
- A: Overall hydrogen-bond acidity
- B: Overall hydrogen-bond basicity
- V: McGowan's Characteristic Volume
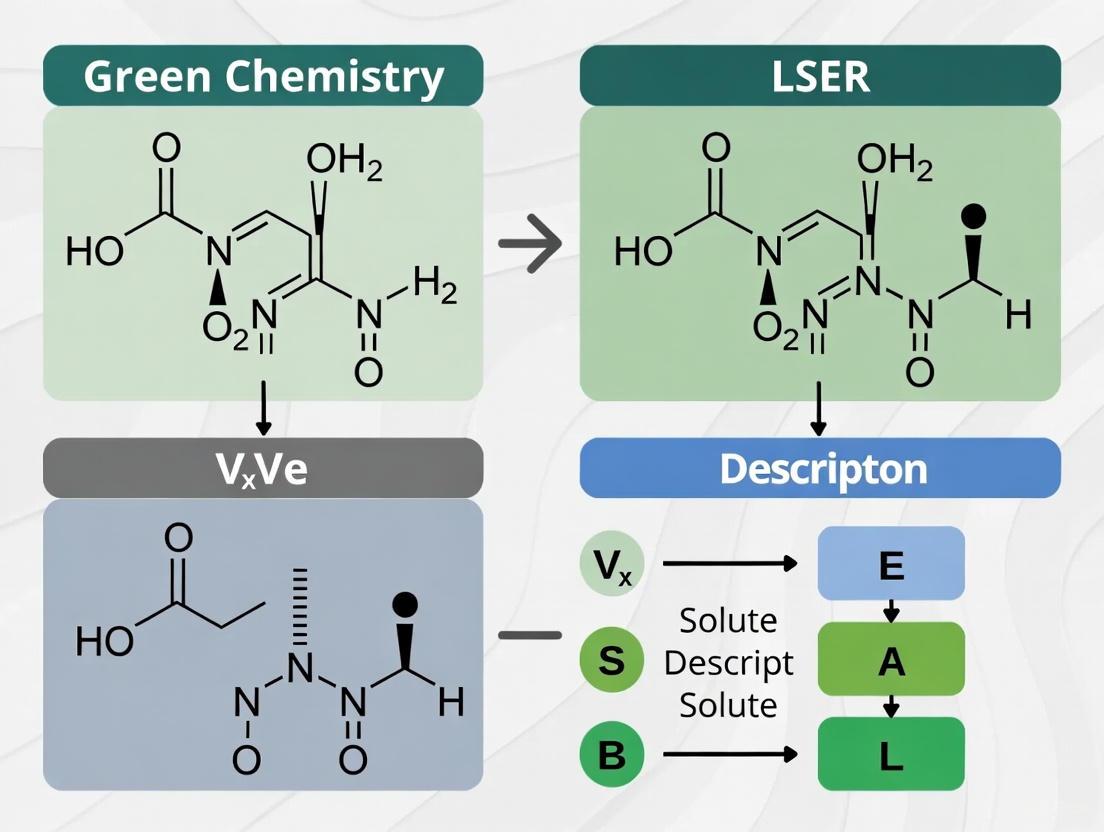
The lowercase letters (e, s, a, b, v) are the system coefficients that characterize the complementary effect of the solvent or phase on solute-solvent interactions [1]. Among these descriptors, McGowan's Characteristic Volume (V or Vx) serves as a fundamental parameter representing the molecular size and contributing to the cavity formation energy required to accommodate a solute molecule within a solvent matrix [1]. This guide explores the theoretical foundation, computational determination, experimental assessment, and practical applications of Vx within modern chemical research and drug development.
Theoretical Foundation and Mathematical Formulation
Definition and Physical Interpretation
McGowan's Characteristic Volume (Vx) is defined as the intrinsic molecular volume, typically expressed in units of dm³·mol⁻¹·100, though it is a dimensionless quantity in the LSER equation [11]. It represents the van der Waals volume of a molecule, corresponding to the space occupied by a molecule that is impenetrable to other molecules at ordinary temperatures [13]. In thermodynamic terms, the Vx descriptor primarily reflects the endothermic cavity formation process and dispersive interactions that occur when a solute is transferred between phases [1]. The product term vV in the LSER equation quantifies the energy required to create a suitably sized cavity in the solvent to accommodate the solute molecule.
Calculation from Molecular Structure
Unlike several other LSER descriptors that require experimental determination, Vx can be calculated directly from molecular structure using a simple algorithm based on atomic contributions and bond counts [11] [14]. The standard calculation method is as follows:
Vx = (Σ Atomic Contributions - Σ Bond Contributions) / 100
Table: Atomic and Bond Contributions for Calculating McGowan's Characteristic Volume
| Component | Contribution Value | Notes |
|---|---|---|
| Carbon Atom | 0.01635 dm³·mol⁻¹ | All carbon types |
| Hydrogen Atom | 0.00110 dm³·mol⁻¹ | All hydrogen types |
| Other Atoms | Element-specific volumes | Based on van der Waals radii |
| Single Bond | -0.00578 dm³·mol⁻¹ | Between any two atoms |
| Double Bond | -0.01156 dm³·mol⁻¹ | Exactly twice single bond value |
| Triple Bond | -0.01734 dm³·mol⁻¹ | Exactly three times single bond value |
The calculation accounts for atomic overlaps in covalent bonding by subtracting bond contributions, resulting in a more accurate representation of the actual molecular volume than simple atomic volume summation [13]. This approach aligns with the concept that van der Waals molecular volume is smaller than the sum of individual atomic volumes due to covalent bond shortening [13].
Diagram: Computational workflow for determining McGowan's Characteristic Volume (Vx) from molecular structure, showing the sequence of summing atomic contributions and adjusting for bond overlaps.
Relationship to Other Molecular Volume Descriptors
Comparison with van der Waals Volume
The van der Waals molecular volume (VvdW) is conceptually similar to Vx but is typically calculated using different methodologies. VvdW is defined as the volume occupied by a molecule that is impenetrable to other molecules, calculated using van der Waals radii of atoms [13]. The calculation involves representing atoms as interlocking spheres with radii corresponding to their van der Waals radii, with the covalent bond distance being shorter than the sum of these radii [13]. While Vx and VvdW both represent intrinsic molecular volumes, Vx employs a simplified calculation method based on atomic contributions and bond corrections, making it more accessible for rapid computation in large-scale chemical database applications [15].
Table: Comparison of Molecular Volume Descriptors
| Descriptor | Basis of Calculation | Units | Key Applications | Advantages |
|---|---|---|---|---|
| McGowan's Vx | Sum of atomic volumes minus bond contributions | dm³·mol⁻¹·100 | LSER models, partition coefficients | Rapid calculation from structure, consistent with LSER framework |
| van der Waals Volume | Sum of atomic spheres with van der Waals radii | ų or nm³ | Molecular modeling, packing calculations | More physically accurate representation of excluded volume |
| Molecular Volume (υ) | Molar volume from molecular weight and density | cm³·mol⁻¹ | Thermodynamic property prediction | Directly measurable, relates to bulk properties |
| Characteristic Volume | McGowan's calculated molecular volume [13] | dm³·mol⁻¹·100 | Chromatographic retention prediction | Specifically parameterized for LSER applications |
McGowan Volume in Context of Steric Descriptors
In modern computational chemistry, Vx represents one of several approaches to quantifying molecular size and steric properties. Contemporary research continues to develop complementary descriptors, such as buried volume and Sterimol parameters, which offer alternative perspectives on steric effects, particularly in catalysis and drug design [16]. The recently developed HeteroAryl Descriptors database (HArD), for instance, includes multiple steric descriptors to capture different aspects of molecular size and shape for heteroaromatic compounds [16]. Despite these advances, Vx remains particularly valuable in LSER applications due to its parameterization within the established solvation parameter model and its computational efficiency.
Experimental Determination and Validation
Chromatographic Methods for Descriptor Assignment
While Vx can be calculated directly from structure, experimental techniques are essential for validating its accuracy and determining other LSER descriptors. The Solvent Method provides a robust multi-technique approach for descriptor assignment [14]. A streamlined experimental design requires 26 total measurements across different techniques [14]:
Table: Experimental Measurements for LSER Descriptor Determination
| Technique | Experimental Conditions | Number of Measurements | Descriptors Validated |
|---|---|---|---|
| Gas Chromatography | 3 retention factor measurements in 60°C range on four columns | 12 measurements | L, S, A, B |
| Reversed-Phase Liquid Chromatography | 3 retention factor measurements in 30% (v/v) acetonitrile composition range on two columns | 6 measurements | S, A, B |
| Liquid-Liquid Partition | Eight partition constant measurements in totally organic and aqueous biphasic systems | 8 measurements | E, S, A, B |
This streamlined approach represents a significant improvement on earlier single-technique approaches, allowing simultaneous determination of E, S, A, B, and L descriptors with minimal bias compared to established database values [14]. For Vx specifically, calculation from structure remains the preferred method, with experimental retention data serving to validate its accuracy in the context of overall LSER model performance.
Substantial efforts have been dedicated to creating comprehensive, validated databases of LSER descriptors. The Wayne State University (WSU) descriptor database represents one of the most authoritative sources, recently updated to the WSU-2025 version containing descriptors for 387 varied compounds [11]. This expanded and updated database provides improved precision and predictive capability compared to its predecessors, with Vx values calculable for all entries [11]. Computational chemistry toolkits such as the Chemistry Development Kit (CDK) implement algorithms for calculating Vx and related volume descriptors, enabling high-throughput screening of compound libraries [15].
Applications in Pharmaceutical Research and Environmental Chemistry
Drug Discovery and Bioavailability Prediction
In pharmaceutical research, Vx serves as a critical parameter for predicting absorption, distribution, metabolism, and excretion (ADME) properties. The descriptor contributes to models of lipophilicity and membrane permeability, which directly influence drug bioavailability. In reversed-phase liquid chromatography, which simulates biomembrane partitioning, the Vx term directly influences retention factor values through its relationship to cavity formation energy [13]. Research on Per- and Polyfluoroalkyl Substances (PFAS) binding to human serum albumin (HSA) has demonstrated the significance of molecular volume descriptors in predicting bioaccumulation potential and protein interaction affinities [17].
Environmental Fate Modeling
Vx is particularly valuable in environmental chemistry for predicting the partitioning behavior of organic contaminants between different environmental compartments. The descriptor features prominently in models predicting air-water, soil-water, and sediment-water partition coefficients, which are essential for understanding the transport, persistence, and ecological impact of pollutants. The solvation parameter model, with Vx as a key component, has been successfully applied to predict the environmental distribution of diverse chemical classes, from hydrocarbons to complex industrial chemicals [1].
Chromatographic Retention Prediction
In analytical chemistry, Vx contributes significantly to accurate retention prediction in various chromatographic modes, including reversed-phase liquid chromatography and gas chromatography [12]. The characteristic volume descriptor helps characterize the hydrophobic contribution to retention, complementing polar and hydrogen-bonding interactions captured by other LSER parameters. Recent research has explored fast characterization methods based on the Abraham solvation parameter model for both reversed-phase and hydrophilic interaction liquid chromatography (HILIC), with Vx playing a consistent role in retention models [12].
Current Research and Emerging Applications
Integration with Equation-of-State Thermodynamics
Recent research has explored the interconnection between LSER descriptors and equation-of-state thermodynamics through the development of Partial Solvation Parameters (PSP) [1]. This approach aims to extract thermodynamic information from the LSER database and facilitate its transfer to other molecular thermodynamics applications. The Vx descriptor contributes to the estimation of dispersion interactions within this framework, helping to bridge QSPR-type databases with equation-of-state developments [1].
Expansion to Heteroaromatic Compound Space
The development of specialized databases for heteroaryl substituents represents an important advancement in steric descriptor applications. The recently introduced HeteroAryl Descriptors database (HArD) comprises DFT-computed steric and electronic descriptors for over 31,500 heteroaryl substituents [16]. While including alternative steric parameters like buried volume and Sterimol parameters, such databases complement the Vx descriptor by providing specialized characterization of important pharmaceutical scaffolds [16].
Machine Learning and Predictive Modeling
Vx continues to serve as a fundamental feature in quantitative structure-activity relationship (QSAR) and machine learning models for chemical property prediction. Its computational efficiency and physical interpretability make it particularly valuable for large-scale virtual screening and chemical priority setting. Recent studies on PFAS-protein interactions have demonstrated how volume-related descriptors like the packing density index (PDI), defined as the ratio between McGowan volume and total surface area, can provide insights into binding affinity and toxicity mechanisms [17].
Research Reagent Solutions
Table: Essential Computational and Experimental Resources for McGowan Volume Research
| Resource/Tool | Type | Function/Application | Key Features |
|---|---|---|---|
| WSU-2025 Descriptor Database [11] | Database | Reference data for LSER parameters | 387 compounds with validated descriptors; updated values |
| Solver Method [14] | Methodology | Experimental descriptor assignment | Multi-technique approach (GC, LC, partition) |
| Chemistry Development Kit (CDK) [15] | Software Library | Molecular descriptor calculation | Open-source, includes Vx implementation |
| MarvinSketch [13] | Software Application | van der Waals volume and area calculation | Commercial implementation with graphical interface |
| HArD Database [16] | Specialized Database | Steric descriptors for heteroaryl groups | >31,500 heteroaryl substituents with DFT-computed parameters |
| Abraham Solvation Parameter Model [1] | Theoretical Framework | Prediction of partition and solubility properties | Established LSER equations with system-specific coefficients |
Linear Solvation Energy Relationships (LSERs) represent a powerful quantitative approach for predicting a wide array of physicochemical and biological properties based on the molecular structure of a compound. The foundational LSER model, as developed by Abraham, is described by the following equation:
Property = c + vVx + eE + sS + aA + bB + lL
In this model, the capital letters (Vx, E, S, A, B, L) are the solute descriptors, each quantifying a specific aspect of a molecule's potential for intermolecular interactions. The lower-case letters (c, v, e, s, a, b, l) are the system coefficients, determined via regression analysis, which reflect the property's sensitivity to each interaction type in a given system. The E descriptor, officially termed the excess molar refractivity, is a central parameter in this framework. It serves as a combined measure of a molecule's polarizability and its capacity to participate in dispersion interactions. Unlike simple refractive index measurements, the E descriptor is specifically constructed to be largely independent of strong, specific interactions like hydrogen bonding, making it a unique and fundamental property in LSER studies for predicting solubility, partitioning behavior, and pharmacokinetic properties.
Theoretical Foundation of Excess Molar Refractivity
Defining Excess Molar Refractivity
The excess molar refractivity (E) is derived from the Lorenz-Lorentz equation, which relates the refractive index of a substance to its molar mass and density. The molar refractivity (R) of a compound is given by:
R = (n² - 1)/(n² + 2) × (M / ρ)
Where:
- n is the refractive index
- M is the molar mass
- ρ is the density
The excess molar refractivity is then defined as the difference between the compound's molar refractivity and the molar refractivity of a hypothetical hydrocarbon of the same molecular volume. This "excess" quantifies the contribution from π- and n-electrons to the overall polarizability, which is why it is also considered a measure of the solute's polarizability and dispersion interaction capability. In the context of LSERs, the E descriptor is a dimensionless quantity, typically normalized and determined from experimental chromatographic or partition data.
Physical Interpretation: Polarizability and Dispersion Forces
The E descriptor fundamentally captures a molecule's ability to undergo electronic polarization—the temporary distortion of its electron cloud in response to an electric field, such as that generated by a nearby molecule. This induced dipole moment is the origin of dispersion (London) forces, which are universal attractive forces present between all atoms and molecules.
- Polarizability: A higher value of the E descriptor indicates a larger, more easily deformable electron cloud. This is typically associated with molecules containing conjugated π-systems, aromatic rings, and heavy atoms (e.g., sulfur, bromine, iodine).
- Dispersion Interactions: In solution, these induced dipole-induced dipole interactions are a major component of the solvation energy. A solute with a high E value will generally experience stronger dispersion interactions with non-polar or polarizable solvents and phases. In biological systems, these interactions are critical for drug binding to hydrophobic pockets in proteins and for passive diffusion through lipid membranes.
Experimental Determination and Methodologies
Direct Measurement via Refractometry
The most straightforward method for determining molar refractivity, and by extension informing the E descriptor, involves measuring the refractive index (n) and density (ρ) of a liquid solute.
Table 1: Key Instrumentation for Direct Refractivity Measurements
| Instrument | Measured Property | Brief Principle of Operation | Key Considerations |
|---|---|---|---|
| Abbé Refractometer | Refractive Index (n) | Measures the critical angle of total internal reflection for a liquid sample. | Requires only a small sample volume; standard method for n. |
| Digital Density Meter | Density (ρ) | Measures the natural oscillation frequency of a U-shaped glass tube filled with the sample (e.g., Anton Paar densimeter). | Provides high-precision density data; often thermostatted. |
Experimental Protocol:
- Sample Preparation: Ensure the compound of interest is pure and in liquid form. If solid, it may need to be dissolved in a solvent, though this complicates the calculation.
- Density Measurement: Fill the densimeter cell with the sample and record the density (ρ) at a controlled temperature (e.g., 25.0 °C). The instrument is typically calibrated with air and water.
- Refractive Index Measurement: Place a drop of the sample on the prism of the refractometer and record the refractive index (n) at the same controlled temperature.
- Calculation: Use the measured n and ρ values, along with the known molar mass (M), to calculate the molar refractivity (R) using the Lorenz-Lorentz equation.
Chromatographic Determination of the E Descriptor
For compounds that are not readily available as pure liquids, or for high-throughput determination, reversed-phase liquid chromatography (RPLC) is a powerful and common indirect method. The retention factor (log k) in a chromatographic system correlates with the LSER descriptors.
Experimental Protocol for RPLC-Derived E Values:
- Chromatographic System Setup:
- Column: A non-polar stationary phase is used, such as an octadecylsilyl (ODS or C18) column (e.g., Purosphere RP-18e) [18].
- Mobile Phase: A binary mixture of water and a water-miscible organic modifier (e.g., methanol, acetonitrile) is used.
- Detection: A UV-Vis or refractive index detector is employed.
- Calibration: A set of reference compounds with known E descriptor values is run isocratically at different mobile phase compositions. The retention factor, log k, is calculated for each compound: k = (tR - t0) / t0, where tR is the compound's retention time and t0 is the column void time.
- Measurement: The compound of unknown E is run under the same chromatographic conditions, and its log k is measured.
- Correlation and Prediction: A multi-linear regression is performed on the calibration set data to establish the system coefficients for the LSER equation in that specific chromatographic system. The established model is then used to back-calculate the unknown solute's E descriptor from its measured log k.
Quantitative Data and Property Correlations
The E descriptor's utility is demonstrated by its strong correlation with numerous physicochemical properties. The following table summarizes key relationships observed in research.
Table 2: Correlations of the E Descriptor with Physicochemical and Biological Properties
| Property / System | Correlation with E | Interpretation & Application | Representative Study |
|---|---|---|---|
| Octanol-Water Partition Coefficient (log P) | Positive | Higher polarizability favors partitioning into the organic (octanol) phase due to enhanced dispersion interactions. | Foundational LSER studies |
| Blood-Brain Barrier Permeability (log BB) | Positive | Compounds with greater polarizability diffuse more readily through lipid membranes of the BBB, a key consideration in CNS drug development [18]. | QSAR studies on drug candidates [18] |
| Chromatographic Retention (log k) on Non-polar Phases | Positive | Increased dispersion interactions with the alkyl chain (C18) stationary phase lead to longer retention times. | RPLC method development |
| Aqueous Solubility | Generally Negative (for non-ionic compounds) | Strong dispersion interactions in the aqueous phase are unfavorable; high-E compounds are "squeezed out" into a non-aqueous phase. | Solubility prediction models |
Research on poly(ethylene glycol)s (PEGs) and their aqueous solutions further illustrates the role of intermolecular interactions, including those captured by the E descriptor. Studies measuring properties like excess molar volume (VmE) and deviation in viscosity (Δη) provide insights into the disruption of water's hydrogen-bonding network and the formation of new glycol-water H-bonds, which are influenced by the polarizability and size of the solute molecules [19].
The Scientist's Toolkit: Essential Reagents and Materials
Successful experimental determination of parameters related to excess molar refractivity relies on specific reagents and instrumentation.
Table 3: Key Research Reagent Solutions and Materials
| Item / Reagent | Function / Purpose | Example & Notes |
|---|---|---|
| ODS (C18) Chromatography Column | Stationary phase for reversed-phase HPLC; provides a non-polar environment for measuring partitioning behavior. | Purosphere RP-18e [18]; the gold standard for log P and LSER descriptor determination. |
| Immobilized Artificial Membrane (IAM) Column | Biomimetic stationary phase that mimics cell membranes; used for predicting pharmacokinetic properties like BBB permeability [18]. | IAM.PC.DD2 columns are used to study passive drug transport. |
| HPLC-Grade Solvents | Mobile phase components for creating a consistent elution environment in chromatography. | Acetonitrile, Methanol, and High-purity Water (e.g., from a Milli-Q system). |
| Reference Compound Sets | Calibrants with known LSER descriptors for establishing system coefficients in chromatographic methods. | Sets often include simple aromatics, alkanes, and compounds with various functional groups. |
| Digital Refractometer | Instrument for direct, precise measurement of a solution's refractive index (n). | Abbé or automated digital refractometers. |
| Vibrating-Tube Density Meter | Instrument for high-precision density (ρ) measurements of liquids and solutions. | Anton Paar digital densimeter, used in studies of aqueous PEG solutions [19]. |
Applications in Pharmaceutical Research and Drug Development
The E descriptor is particularly valuable in Quantitative Structure-Activity Relationship (QSAR) and Quantitative Structure-Property Relationship (QSPR) modeling, which are cornerstones of modern drug discovery.
Predicting Blood-Brain Barrier (BBB) Permeability: The ability of a drug candidate to cross the BBB is crucial for central nervous system targets. Research has established that BBB penetration is promoted by high lipophilicity and a weak hydrogen bonding potential [18]. The E descriptor, representing favorable dispersion interactions with the lipid-rich membrane, is a key positive contributor in LSER models for log BB (where log BB = log(Cbrain/Cblood)) [18]. Models using chromatographic retention data from ODS and IAM columns to predict log BB rely heavily on an accurate determination of the E descriptor.
Solubility and Absorption Prediction: During the early stages of drug design, predicting a molecule's aqueous solubility and intestinal absorption is vital. The E descriptor contributes to models that predict these properties by accounting for the energy cost of cavity formation in water and the dispersion interactions that drive partitioning into biological membranes.
Within the comprehensive LSER framework, the excess molar refractivity (E) descriptor is an indispensable tool for quantifying the polarizability and dispersion interaction capacity of a molecule. Its determination, whether through direct physico-chemical measurements or indirect chromatographic methods, provides critical insights that drive predictive modeling in environmental chemistry and pharmaceutical sciences. As drug discovery efforts increasingly rely on in silico methods to prioritize synthetic targets, the accurate determination and application of the E descriptor and its fellow LSER parameters (Vx, S, A, B, L) will remain a fundamental aspect of rational molecular design, enabling researchers to optimize key properties like membrane permeability and bioavailability more efficiently.
Linear Solvation Energy Relationships (LSERs) utilize a set of solute descriptors, commonly known as the Abraham descriptors, to quantitatively predict physicochemical properties and biological activities. These descriptors are encapsulated in the acronym Vx E S A B L, where each letter represents a specific molecular property: Vx is McGowan's characteristic volume, E is the excess molar refractivity, S represents dipolarity/polarizability, A denotes hydrogen-bond acidity, B signifies hydrogen-bond basicity, and L is the gas-hexadecane partition coefficient. The S descriptor specifically quantifies a molecule's ability to engage in dipole-dipole and dipole-induced dipole interactions. It measures a compound's polarity and polarizability, representing the solute's effective tendency to stabilize itself through nonspecific interactions with polar solvents. In pharmacological contexts, the S descriptor helps predict solubility, permeability, and membrane transport properties, as these processes fundamentally depend on molecular interactions in various biological environments. Accurate determination of S is therefore crucial for rational drug design, particularly in optimizing absorption, distribution, and bioavailability characteristics of lead compounds.
Theoretical Foundations of Dipolarity and Polarizability
Electric Dipole Moments in Molecular Systems
An electric dipole moment arises from the separation of positive and negative charges within a molecular system. For the simplest case of two point charges +q and -q separated by distance vector d, the dipole moment μ is defined as μ = qd. In molecular systems, permanent dipole moments exist in neutral molecules due to unequal electron distribution between atoms of different electronegativities. The magnitude of this permanent dipole significantly influences how a molecule interacts with its environment, particularly in condensed phases and biological systems. When a molecule with a permanent dipole moment is placed in an electric field, the field exerts a torque that tends to align the dipole with the field direction, while thermal motion tends to randomize the orientation. This competition between ordering and disordering forces governs many dielectric properties of materials and contributes to the S descriptor in LSER formulations. The energy of interaction between a permanent dipole and an external electric field is given by U = -μ·E, which forms the basis for understanding orientation polarization in dielectric materials. [20]
Polarization Mechanisms and Polarizability
Polarizability (α) describes how easily the electron cloud of a molecule can be distorted by an external electric field, leading to an induced dipole moment (μ_ind = αE). This induced moment exists only while the field is applied and contributes to the overall polarization of the substance. The relationship between molecular polarizability and the S descriptor is fundamental to LSER theory, as it captures the nonspecific, non-directional interactions between solute and solvent molecules. Several polarization mechanisms contribute to a molecule's overall response to electric fields: Electronic polarization involves displacement of the electron cloud relative to the nuclei, atomic polarization involves relative displacement of atomic nuclei within the molecule, and orientation polarization involves alignment of permanent dipoles with the field. For drug-like molecules, the electronic polarizability often correlates with π-electron systems and aromatic character, making it particularly relevant for pharmaceutical compounds containing aromatic rings or conjugated systems. The S descriptor effectively integrates these various polarization contributions into a single parameter that describes a molecule's overall tendency to engage in dipole-related interactions. [20]
Interfacial Effects on Dipole Behavior
When molecules are situated near interfaces, as commonly occurs in biological systems and chromatography, their dipole moments and polarization characteristics can be significantly modified. The presence of an interface creates a dielectric discontinuity that affects both the inducing field and the radiation emitted by the oscillating dipole. Research has shown that the electric dipole moment of a particle near an interface can be described by resolvent functions Υ∥(h) and Υ⊥(h) that depend on the dimensionless distance h between the particle and the interface. These functions exhibit resonance features due to the back-action mechanism where dipole radiation reflects at the interface and modifies its own source. This phenomenon is particularly relevant for understanding molecular behavior at biological membranes, protein surfaces, and stationary phases in chromatographic systems used to determine LSER descriptors. The power emitted by the particle depends on h due to interference between source radiation and reflected radiation, creating an additional distance dependence that shows resonance peaks under certain conditions. [20]
Experimental Methodologies for Dipole Moment Determination
Polarization Laser Spectroscopy
Polarization laser spectroscopy represents a sophisticated approach for measuring electric dipole moments between degenerate quantum states. This method exploits the strong polarization dependence of atomic photo-excitation behavior in a controlled vacuum environment. The experimental protocol involves a two-step resonance excitation process with two laser beams, where precise control of laser polarizations enables different excitation conditions within the same excitation scheme. [21]
Table 1: Key Parameters in Polarization Laser Spectroscopy for Dipole Moment Measurement
| Parameter | Specification | Function |
|---|---|---|
| Laser System | Tunable narrow-bandwidth lasers | Provides precise excitation energy for resonant transitions |
| Vacuum Chamber | Ultra-high vacuum (≤10⁻⁸ mbar) | Elimates collisional broadening and quenching |
| Polarization Control | Linear, circular, or elliptical polarizers | Creates specific excitation conditions for quantum states |
| Detection System | Fluorescence or ionization detectors | Measures population transfer between quantum states |
| Quantum Mechanical Model | Includes μ as fitting parameter | Extracts dipole moment from experimental data |
The experimental workflow begins with atomization of the sample in the vacuum chamber, typically using high-temperature heating or laser ablation. Two independently tunable laser systems with precise polarization control are then employed for sequential excitation of the target quantum states. The first laser prepares atoms in an intermediate state, while the second laser promotes them to the final excited state. By systematically varying the polarizations of both lasers and measuring the resulting excitation curves, researchers can obtain sufficient data to extract the transition dipole moment through quantum mechanical fitting. This method has been successfully applied to uranium atom transitions, revealing dipole moments of 0.16 and 4.1 Debye for specific transitions that had not been previously measured directly. [21]
Electric Field-Induced Alignment Methods
Another approach for determining molecular dipole moments involves observing the alignment or orientation of molecules in external electric fields. Stark spectroscopy applies a controlled electric field to a molecular sample and measures the resulting shifts and splittings in rotational or vibrational spectra. The magnitude of these effects depends directly on the permanent dipole moment, allowing for precise quantification. For symmetric top molecules, the Stark effect produces characteristic splitting patterns that can be analyzed to determine both the magnitude and orientation of the dipole moment vector relative to the principal molecular axes. Modern implementations of this technique often combine supersonic jet expansion with high-resolution microwave or infrared spectroscopy, enabling the study of isolated molecules with minimal thermal broadening.
Computational Approaches for S Descriptor Determination
While experimental methods provide direct measurements of dipole moments, computational approaches offer efficient means for estimating the S descriptor for LSER applications. Quantum mechanical calculations, particularly density functional theory (DFT), can predict molecular dipole moments and polarizabilities with reasonable accuracy. These calculations typically involve geometry optimization followed by property evaluation at the equilibrium structure. For the S descriptor specifically, chromatographic methods using well-characterized stationary phases provide experimental determination through solvation parameter models. The S value is derived from the difference in retention behavior between polar and nonpolar stationary phases, capturing the molecule's overall dipolarity and polarizability characteristics that govern its interactions in biological and environmental systems.
Research Reagent Solutions for Dipole Moment Studies
Table 2: Essential Research Reagents for Dipole Moment and Polarization Experiments
| Reagent/Equipment | Function | Technical Specifications |
|---|---|---|
| Tunable Dye Laser System | Provides precise excitation wavelengths | Spectral resolution <0.1 cm⁻¹, wavelength range 250-1000 nm |
| Ultra-High Vacuum Chamber | Creates collision-free environment for spectroscopy | Base pressure ≤10⁻⁸ mbar, with sample introduction system |
| Electro-Optic Modulators | Controls polarization states of laser beams | Modulation frequency >100 kHz, extinction ratio >1000:1 |
| Quantum Chemistry Software | Computes molecular electronic properties | DFT functionals (B3LYP, ωB97X-D), basis sets (cc-pVDZ, aug-cc-pVTZ) |
| High-Voltage Stark Electrodes | Generates uniform electric fields for alignment | Field strength up to 100 kV/cm, parallel plate configuration |
| Supersonic Jet Expansion Source | Cools molecules for high-resolution spectroscopy | Backing pressure 1-10 bar, pulsed valve operation |
Visualization of Experimental Workflows
Polarization Spectroscopy Methodology
LSER S Descriptor Determination Framework
Data Presentation and Comparative Analysis
Experimental Dipole Moment Measurements
Table 3: Experimentally Determined Electric Dipole Moments for Selected Transitions
| Atomic/Molecular System | Transition Energy (cm⁻¹) | Dipole Moment (Debye) | Measurement Method |
|---|---|---|---|
| Uranium Atom | 16,900 → 33,939 | 0.16 | Polarization Laser Spectroscopy |
| Uranium Atom | 16,900 → 34,599 | 4.10 | Polarization Laser Spectroscopy |
| Representative Drug Molecule | N/A | 1.5-4.5 | Stark Spectroscopy |
| Polar Aromatic Compound | N/A | 3.0-5.0 | Computational DFT Methods |
The data presented in Table 3 illustrates the range of dipole moments measurable with current techniques. The significant difference between the two uranium transitions (0.16 vs. 4.10 Debye) highlights the substantial variation that can exist even within the same atomic system. For pharmaceutical compounds, dipole moments typically fall in the 1.5-5.0 Debye range, with higher values often correlating with improved aqueous solubility but potentially reduced membrane permeability. [21]
Correlation Between Dipole Properties and S Descriptor
Table 4: Relationship Between Molecular Properties and LSER S Descriptor
| Molecular Characteristic | Effect on S Descriptor | Impact on Solvation Properties |
|---|---|---|
| Large Permanent Dipole Moment | Increases S value | Enhanced solubility in polar solvents |
| High Electronic Polarizability | Increases S value | Stronger dispersion interactions |
| Conjugated π-Systems | Significantly increases S | Improved interaction with aromatic stations |
| Polar Functional Groups | Increases S | Better hydration and polar solvation |
| Nonpolar Aliphatic Chains | Decreases S | Increased hydrophobicity |
The S descriptor integrates various dipole-related properties into a single parameter that effectively predicts solvation behavior across different media. Understanding these relationships enables researchers to rationally design compounds with optimized distribution characteristics for pharmaceutical applications. Molecules with balanced S values typically exhibit improved drug-like properties, adequate for both aqueous solubility and membrane permeation.
Hydrogen-bond (H-bond) acidity and basicity are fundamental molecular properties that quantify a substance's capacity to act as a hydrogen-bond donor (HBD) or acceptor (HBA), respectively. Within the framework of Linear Solvation Energy Relationships (LSERs), these are formally defined as the solute descriptors A (hydrogen-bond acidity) and B (hydrogen-bond basicity) [22]. They are integral components of the Abraham solvation parameter model, which expresses a solute's property (such as a partition coefficient) as a linear combination of its molecular descriptors: Vx, E, S, A, B, and L [23]. The A parameter represents the solute's ability to donate a hydrogen bond, while the B parameter represents its ability to accept one [22]. The accurate determination of these parameters is critical for researchers and drug development professionals, as they allow for the prediction of a molecule's behavior in different environments, influencing solubility, permeability, bioavailability, and binding affinity [24] [25] [23].
Experimental Scales and Measurement Methodologies
Several experimental scales have been developed to quantify hydrogen-bond strength, primarily based on measuring equilibrium constants for complex formation in poorly coordinating solvents.
The pK₍BHX₎ Scale for Hydrogen-Bond Basicity
The pKBHX scale is a widely used measure of hydrogen-bond acceptor strength. It is defined as the base-10 logarithm of the equilibrium constant (K) for the 1:1 complex formation between a hydrogen-bond acceptor and the reference donor 4-fluorophenol in carbon tetrachloride [24] [26]. Under these conditions, pKBHX values for common organic functional groups span approximately six orders of magnitude, typically ranging from -1 for weak acceptors like alkenes to 5 for strong acceptors like amides and N-oxides [24] [26].
Table 1: Experimentally Measured pKBHX Values for Representative Functional Groups
| Functional Group | Typical pK₍BHX₎ Range | Representative Example |
|---|---|---|
| Alkene | -1 to 0 | --- |
| Amine | ~1.4 (varies with sterics) | Triisopropylamine: 0.30 [24] |
| Amide | 2.0 to 2.5 | --- |
| Carbonyl | ~2.0 (varies with substitution) | --- |
| N-Oxide | >3.0 | --- |
The Abraham Scale and the ln K₍eq₎ Measurement
Abraham's A and B parameters are determined from the log₁₀K values for hydrogen bond formation between acids and bases in inert solvents like CCl₄ [22]. An alternative, experimentally accessible approach for measuring donor strength uses a colorimetric pyrazinone sensor.
Experimental Protocol: UV-Vis Titration for Hydrogen-Bond Donor Strength
- Principle: A pyrazinone sensor undergoes a colorimetric shift upon complexation with a hydrogen-bond donor. The binding constant (Keq), determined by titration, is a direct measure of the analyte's hydrogen-bond donating ability, reported as lnKeq [25].
- Procedure:
- Environment: Titrations are performed in dichloromethane to limit confounding non-covalent interactions and amplify hydrogen bonding effects [25].
- Measurement: The analyte is titrated into a solution of the pyrazinone sensor.
- Analysis: The resulting UV-Vis spectral shifts are measured, and the binding constant (Keq) is calculated from the titration data. A larger lnKeq value corresponds to a stronger hydrogen-bond donor [25].
- Application: This method is suitable for weak to moderate donors, including various N-H and O-H containing compounds such as heterocycles, amides, sulfonamides, and alcohols that are common in pharmaceuticals [25].
Table 2: Experimentally Determined Hydrogen-Bond Donor Strengths (ln K₍eq₎) for Selected Motifs
| Compound Class | Example Structure | ln K₍eq₎ | Notes |
|---|---|---|---|
| Aliphatic Alcohols | Compound 44 / 45 | 0.86 / 0.94 | Very weak donors [25] |
| Benzyl Alcohol | Compound 50 | 1.93 | Stronger than aliphatic due to inductive effects [25] |
| Amines | Compound 1 | ~2.0 (weak) | Among the weakest successful titrations [25] |
| Primary Amide | Compound 13 | >2.41 | Stronger than secondary amides [25] |
| Imidazole | Unsubstituted | 3.42 | Strength decreases with alkyl substitution [25] |
| Imides | Compound 22 | Relatively strong | Enhanced by two electron-withdrawing carbonyls [25] |
Computational Prediction of A and B Parameters
Computational methods provide an efficient path to predicting hydrogen-bonding strength, avoiding laborious experimental measurements.
Prediction from Electrostatic Potential (pK₍BHX₎)
A robust black-box workflow for predicting site-specific hydrogen-bond basicity (pKBHX) uses the minimum electrostatic potential (Vmin) in the region of a hydrogen-bond acceptor's lone pairs [24] [26].
Computational Protocol: Vmin-Based pK₍BHX₎ Prediction
- Step 1: Conformer Generation. A conformer search is run on the input molecule using the ETKDG algorithm in RDKit, followed by MMFF94 optimization [24].
- Step 2: Conformer Filtering. The conformational ensemble is filtered using the CREST screening protocol and GFN2-xTB energies to remove duplicates and high-energy conformers [24].
- Step 3: Neural Network Optimization. The remaining conformers are scored and optimized with the AIMNet2 neural network potential, with the lowest-energy conformer selected for the final calculation [24].
- Step 4: DFT Single-Point Calculation. A single density-functional-theory (DFT) calculation at the r2SCAN-3c level is performed on the optimized geometry to compute the electrostatic potential [24].
- Step 5: Locate Vmin and Scale. The electrostatic potential minima (Vmin) near each hydrogen-bond accepting atom are located by numerical optimization. These values are linearly scaled using functional-group-specific parameters to predict experimental pKBHX values [24]. This workflow achieves a mean absolute error of about 0.19 pKBHX units across diverse functional groups [24].
Figure 1: Computational workflow for predicting hydrogen-bond basicity (pKBHX) from the electrostatic potential.
Prediction from Atomic Properties and Quantum Chemical Topology
The Abraham A parameter correlates strongly with the computed partial charge on the most positive hydrogen atom in the molecule, though steric effects can also play a significant role [22]. In contrast, the S parameter, which represents polarity/polarizability, correlates with the molecular dipole moment, the partial charge on the most negative atom, and for single-ring aromatics, the molecular polarizability [22].
A Quantum Chemical Topology (QCT) approach has also shown success, linking experimentally measured pKBHX values to the change in the atomic energy of the hydrogen atom, ΔE(H), upon complexation. This method has achieved strong correlations for several common HBDs, including water (r² = 0.96), methanol (r² = 0.95), and 4-fluorophenol (r² = 0.91) [23].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Computational Tools for H-Bond Strength Analysis
| Item / Reagent | Function / Application | Context & Rationale |
|---|---|---|
| 4-Fluorophenol | Reference Hydrogen-Bond Donor | Standard donor for experimental pK₍BHX₎ measurement in CCl₄ [24] [26]. |
| Carbon Tetrachloride (CCl₄) | Inert Solvent | Used in equilibrium constant measurements to minimize solvent interference [24] [22]. |
| Pyrazinone Sensor | Colorimetric H-Bond Acceptor | Enables H-Bond donor strength (ln K₍eq₎) quantification via UV-Vis titration [25]. |
| RDKit | Open-Source Cheminformatics | Used for initial conformer generation with the ETKDG algorithm [24]. |
| AIMNet2 | Neural Network Potential | Accelerates geometry optimization, replacing costly DFT optimizations [24]. |
| r2SCAN-3c Functional | Density Functional Theory (DFT) | Provides a low-cost, high-accuracy method for the final electrostatic potential calculation [24]. |
Applications in Rational Molecular Design and Drug Discovery
Quantifying A and B parameters is not merely an academic exercise; it provides powerful insights for rational molecular design, particularly in medicinal chemistry.
Figure 2: The influence of hydrogen-bond donor and acceptor strength on key physicochemical and pharmacological properties of drug molecules.
A compelling case study from AstraZeneca illustrates this principle. During the optimization of IRAK4 inhibitors, researchers observed that a seemingly minor change—moving a nitrogen atom within a pyrrolopyrimidine scaffold—increased the hydrogen-bond acceptor strength (pKBHX) of two key sites by 0.61 and 0.15 units, respectively. This 4-fold increase in basicity for one site led to decreased lipophilicity, lower membrane permeability, and a higher efflux ratio, all undesirable for an orally bioavailable drug. In contrast, switching to a pyrrolotriazine scaffold lowered the pKBHX values, resulting in more favorable permeability and efflux profiles [26]. This demonstrates how quantitative prediction of hydrogen-bond basicity can directly guide scaffold selection and property-based design.
Linear Solvation Energy Relationships (LSERs) are a powerful tool for predicting the partitioning behavior of solutes across different chemical and biological systems. The widely accepted Abraham model represents this relationship as SP = c + eE + sS + aA + bB + vV, where the dependent variable (SP) is a free-energy-related property, such as the logarithm of a partition coefficient [2]. The capital letters (E, S, A, B, V) are solute-specific descriptors that quantify a molecule's potential for various intermolecular interactions [2]. Within this framework, the gas-hexadecane partition coefficient, denoted as L, serves as a critical solute descriptor. It is defined as the logarithm of the hexadecane-air partition coefficient (log K_hexadecane/air) and is a principal measure of a solute's capability for non-specific van der Waals interactions and its intrinsic lipophilicity [27]. Its prominence stems from its role in characterizing the balance between cavity formation in the condensed phase and the exoergic solute-solvent attractive forces that govern partitioning from the gas phase [2]. The L descriptor is utilized in models predicting partition processes involving a gas phase and a condensed phase, and a modified version of the Abraham equation uses L for all partitioning processes, offering advantages in thermodynamic consistency and for compounds where alternative descriptors like E are difficult to measure [27].
Theoretical Foundation of theLDescriptor
Physico-Chemical Interpretation
The gas-hexadecane partitioning process, characterized by L, is fundamentally governed by two opposing thermodynamic contributions. First, an endoergic process involves creating a cavity of suitable size within the highly structured hexadecane solvent to accommodate the solute molecule. This process requires energy to disrupt the cohesive forces between hexadecane molecules. Second, an exoergic process involves the establishment of attractive, non-specific forces between the solute and the hexadecane solvent molecules once the solute is in the cavity [2]. Hexadecane, a long-chain, non-polar alkane, is an excellent model for biological lipids and inert organic phases. Its primary interactions with solutes are dispersion (London) forces, a type of van der Waals interaction. Therefore, the L value predominantly reflects a solute's polarizability and molecular volume—properties that directly influence the strength of these dispersion interactions [27]. A larger, more polarizable solute will generally experience stronger dispersion forces with hexadecane, leading to a higher L value and indicating greater lipophilicity.
Role in the Goss-Modified Abraham Equation
The central role of the L descriptor is highlighted in the Goss-modified Abraham equation, which provides a unified form for all partition processes:
log Ki,xy = lxy Li + sxy Si + axy Ai + bxy Bi + vxy Vi + cxy [27].
In this equation:
L_iis the solute's hexadecane-air partition coefficient descriptor.- The lower-case system coefficients (
l_xy,s_xy,a_xy,b_xy,v_xy) describe the difference in capacity between phasesxandyfor the respective intermolecular interactions [27].
This formulation offers key advantages. It allows for the direct application of the thermodynamic cycle to convert, for example, a model for partitioning between a phase and air into a model for partitioning between that phase and water [27]. Furthermore, for solid compounds where the refractive index (and thus the E descriptor) cannot be measured, the L value can often be determined experimentally with less error, providing a more reliable descriptor for determining other parameters like S, A, and B [27].
Experimental Determination ofL
Standard Protocol: Inverse Gas Chromatography (IGC)
The established method for determining L values experimentally is Inverse Gas Chromatography (IGC) using non-polar capillary columns [27] [28]. The following protocol details the standard procedure.
Table 1: Key Research Reagents and Materials for IGC Determination of L
| Item Name | Function/Description | Critical Specifications |
|---|---|---|
| SPB-octyl Capillary Column | Non-polar stationary phase; mimics hexadecane partitioning environment. | Chemically bonded octyl polysiloxane film; low column bleed. |
| Hexadecane Calibrants | Set of reference compounds with known L values for calibration. |
Should cover a wide range of L values; high purity (>97%). |
| Test Solute | The compound for which the L value is to be determined. |
High purity (>97%); must be volatile and thermally stable under GC conditions. |
| Carrier Gas | Mobile phase for gas chromatography. | High-purity helium or nitrogen. |
Procedure
- Column Selection and Calibration: A gas chromatograph is equipped with a non-polar capillary column, such as a SPB-octyl column. The column is calibrated using a series of reference compounds with precisely known
Lvalues. The logarithm of the retention factor (log k') for these calibrants is plotted against their knownLvalues to establish a linear calibration curve of the form log k' = c + l L [27]. The retention factork'is calculated ask' = (t_R - t_M) / t_M, wheret_Ris the analyte's retention time andt_Mis the holdup time. - Solute Measurement: The test solute is injected into the GC system, and its retention time is measured under the same isothermal conditions used for calibration.
- Data Analysis: The retention factor (
k') for the test solute is calculated from its measured retention time. TheLvalue is then determined by interpolating thisk'value into the pre-established calibration equation [27].
Critical Experimental Considerations
- Temperature Control: Precise and stable temperature control of the column is essential, as
Lis temperature-dependent. - Column Deactivation: The column must be thoroughly deactivated to eliminate any polar adsorption sites that could interfere with the purely partition-based mechanism [27].
- Solute Suitability: The method is suitable for compounds that are volatile and thermally stable at the operating temperature of the GC. For semi-volatile or thermolabile compounds, alternative methods or predictive tools must be used.
An Alternative Method: Laser Ablation Mass Spectrometry
A more recent and specialized technique for studying partitioning involves laser ablation from a droplet surface coupled with mass spectrometry. While not a direct measurement of L, this method quantitatively characterizes solute partitioning between the bulk liquid and the gas-liquid interface, a process related to hydrophobicity and surface activity [29].
Procedure
- Droplet Deposition: A microliter-sized droplet of the analyte solution is deposited on a metal post.
- Laser Ablation: An infrared laser tuned to the absorption wavelength of the solvent is focused on the droplet surface. Low fluence (energy) settings are used to preferentially ablate material from the surface layer.
- Ionization and Detection: The ablated material is ionized, typically by coalescing with an electrospray ionization plume, and then analyzed by a mass spectrometer.
- Quantification: By holding the droplet volume constant with solvent replenishment and monitoring the ion signal decay over many laser pulses, the data can be fitted to a model based on Langmuir adsorption isotherms to yield quantitative surface partition coefficients [29].
Compendium of ExperimentalLData and Prediction Tools
A wealth of experimental L values has been compiled for environmentally and pharmacologically relevant compounds. These datasets are crucial for developing and validating predictive models.
Table 2: Experimental L Values for Representative Compounds
| Compound Class/Category | Example Compound | Log L (L value) | Experimental Context |
|---|---|---|---|
| Multifunctional Compounds | Environmentally relevant pesticides, drugs, hormones, phthalates | Range: 2.3 to 13.7 | Measured for 104 compounds; standard deviation <0.28 (avg. 0.10) [27]. |
| Environmentally Relevant Compounds | Pesticides, flame retardants, hormones | Range: 4.28 to 15.92 | Measured for 387 compounds to expand pp-LFER applicability [28]. |
Evaluation of Predictive Models forL
For compounds where experimental determination is not feasible, in silico prediction tools are essential. Several software packages have been rigorously evaluated against large experimental datasets.
Table 3: Performance Comparison of L Value Prediction Tools
| Software Tool | Prediction Methodology | Root Mean Squared Error (rmse) | Notable Strengths and Limitations |
|---|---|---|---|
| ABSOLV | Linear solvation energy relationships (LSER) and group contributions. | 0.99 [28] | Performs well for bifunctional compounds but may fail for complex pesticides/drugs [27]. |
| COSMOtherm(X) | Quantum chemistry-based COSMO-RS theory. | 0.94 [28] | Shows the best overall performance; works well for pesticides and drugs [27] [28]. |
| SPARC | Uses mechanistic perturbation theory. | 1.28 [28] | Has problems with highly fluorinated and phosphate-containing compounds [27]. |
| Connectivity Indices | Based on molecular graph topology. | 1.55 [28] | Generally the poorest performance among evaluated tools [28]. |
Visualization of Relationships and Workflows
LSERLDescriptor Meaning and Relationships
The following diagram illustrates the molecular interactions captured by the L descriptor and its relationship to other LSER parameters.
Experimental Workflow for DeterminingL
This flowchart outlines the standard inverse gas chromatography (IGC) protocol for measuring the L value.
The gas-hexadecane partition coefficient L is a fundamental solute descriptor within the LSER framework, providing a precise measure of a solute's capacity for non-specific van der Waals interactions and its intrinsic lipophilicity. Its determination via standardized IGC protocols ensures the generation of high-quality data, which is vital for expanding LSER databases. The ongoing refinement of computational tools like COSMOtherm and ABSOLV is closing the gap between prediction and experiment, enabling reliable estimates of L for complex molecules where measurement is challenging. As a cornerstone parameter, L is indispensable for accurate predictions of environmental transport, biological uptake, and chromatographic retention, thereby playing a critical role in chemical risk assessment and drug development.
Determining and Applying LSER Descriptors: Experimental and Computational Methods in Drug Development
The solvation parameter model is a cornerstone of modern quantitative structure-property relationship (QSPR) studies, providing a robust framework for predicting the behavior of compounds in chemical, biological, and environmental systems [30]. This model utilizes a set of six solute descriptors to characterize molecular interaction capabilities: L (gas-liquid partition coefficient on hexadecane at 298 K), V (McGowan's characteristic volume), E (excess molar refraction), S (dipolarity/polarizability), A (hydrogen-bond acidity), and B (hydrogen-bond basicity) [30] [31]. For certain compounds exhibiting variable hydrogen-bond basicity in aqueous systems, a seventh descriptor (B°) may be employed [32]. These descriptors have become indispensable tools for researchers and drug development professionals seeking to predict partition coefficients, retention factors, and various pharmacokinetic properties without extensive experimental measurement [30] [32].
The theoretical foundation of the model rests on two primary linear free energy relationships (LFERs). For the transfer of a neutral compound from a gas phase to a liquid or solid phase, the model is expressed as log SP = c + eE + sS + aA + bB + lL [2] [1]. For transfer between two condensed phases, the equation becomes log SP = c + eE + sS + aA + bB + vV [32] [1]. In these equations, the system constants (lowercase letters) characterize the intermolecular interactions offered by the specific biphasic system, while the solute descriptors (uppercase letters) quantify the molecule's capability to participate in these interactions [1]. The robustness of this approach lies in its consistency; the solute descriptors are independent of the system and can be used to predict properties in any system for which the constants have been calibrated [30] [32].
Fundamental Solute Descriptors: Definitions and Interpretations
Physical Significance of Each Descriptor
V - McGowan's Characteristic Volume: This descriptor represents the van der Waals volume per mole when molecules are stationary and is a measure of molecular size [32]. It accounts for the energy associated with cavity formation when a compound transfers between two condensed phases [32]. It is easily calculated from molecular structure using the formula: V = [∑(all atom contributions) - 6.56(N - 1 + Rg)]/100, where N is the total number of atoms and Rg is the total number of ring structures [32]. The result is scaled by division by 100 to have similar magnitude to other descriptors.
E - Excess Molar Refraction: This parameter describes a compound's capability to participate in electron lone pair interactions resulting from loosely bound n- and π-electrons [32]. It represents additional dispersion interactions possible for polarizable compounds. For liquids at 20°C, it can be calculated from an experimental refractive index (η) and the compound's characteristic volume: E = 10V[(η² - 1)/(η² + 2)] - 2.832V + 0.528 [32]. The scale is defined so that E = 0 for all n-alkanes.
S - Dipolarity/Polarizability: This descriptor quantifies interactions of a dipole-type that result from a compound's dipolarity and polarizability, representing the combined contribution of orientation and induction interactions [32] [31]. The n-alkanes are assigned a value of zero for this and all other polar interaction descriptors.
A - Overall Hydrogen-Bond Acidity: This parameter describes a compound's hydrogen-bond donating capacity (hydrogen-bond donor strength) [32] [31]. For multifunctional compounds, it represents the summation of hydrogen-bond acidity for all functional groups.
B - Overall Hydrogen-Bond Basicity: This parameter describes a compound's hydrogen-bond accepting capacity (hydrogen-bond acceptor strength) [32] [31]. Certain compounds (e.g., some anilines, alkylamines, sulfoxides) exhibit variable hydrogen-bond basicity in aqueous systems, requiring use of the B° descriptor for reversed-phase liquid chromatography and aqueous-organic partition systems [32].
L - Gas-Hexadecane Partition Coefficient: This descriptor is defined as the logarithm of the gas-liquid partition constant at 25°C with n-hexadecane as the stationary phase [32] [33]. It represents the free energy change arising from dispersion interactions when a compound transfers from an ideal gas phase to n-hexadecane, opposed by the disruption of solvent-solvent interactions required for cavity formation [32].
Table 1: Summary of Abraham Solute Descriptors and Their Determination Methods
| Descriptor | Molecular Property Represented | Primary Determination Methods | Calculability |
|---|---|---|---|
| L | Gas-hexadecane partition coefficient | GC with hexadecane, low-polarity stationary phases | Experimental |
| V | McGowan's characteristic volume | Molecular structure | Calculated from structure |
| E | Excess molar refraction | Refractive index measurement (liquids), estimation (solids) | Calculated for liquids, estimated for solids |
| S | Dipolarity/polarizability | Chromatographic and partitioning methods | Experimental |
| A | Hydrogen-bond acidity | Chromatographic and partitioning methods, NMR spectroscopy | Experimental |
| B/B° | Hydrogen-bond basicity | Chromatographic and partitioning methods | Experimental |
Experimental Determination Methods
Chromatographic Techniques
Chromatographic methods are particularly well-suited for determining solute descriptors because the necessary equipment is available in most laboratories, methods require only small sample amounts, can accommodate impure samples, and have low operating costs [30]. The general approach involves measuring retention factors (log k) in multiple chromatographic systems with known system constants, then solving for the descriptors using the Solver method [32].
Gas Chromatography (GC)
Gas chromatography is highly valuable for determining L and S descriptors, particularly when using low-polarity stationary phases [30] [33]. The determination of the L descriptor requires careful consideration of stationary phase properties. Squalane packed columns and open-tubular columns coated with poly(methyloctylsiloxane) have been identified as effective surrogate systems [33]. Retention on squalane columns is dominated by gas-liquid partitioning with a temperature-dependent contribution from interfacial adsorption [33]. When properly corrected for interfacial adsorption, log L can be estimated to within ±0.026 log units over 60-120°C [33]. For the poly(methyloctylsiloxane) stationary phase, prior knowledge of the solute S descriptor is necessary to avoid significant errors in estimating L for polar compounds [33].
Experimental Protocol for L Descriptor Determination Using GC:
- Select appropriate low-polarity stationary phase (squalane or poly(methyloctylsiloxane)
- Measure retention times for the target compound and reference compounds with known descriptors at multiple temperatures (typically 60-140°C)
- Determine column dead time by injecting air and measuring argon (m/z 40)
- Calculate retention factors: k = (t - t₀)/t₀
- Use system constants previously determined for the GC column to solve for L
- Validate results using compounds with known descriptor values
Recent applications of GC for descriptor determination include characterizing perfluoroalkyl and polyfluoroalkyl substances (PFAS), where four different stationary phases with varying polarities (HP-5ms, DB-200, DB-225ms, SolGel-WAX) were used to determine complete descriptor sets for 47 neutral PFAS [34]. This study revealed the characteristic intermolecular interaction properties of PFAS, such as hydrogen-bonding capabilities influenced by electron-withdrawing perfluoroalkyl groups [34].
Reversed-Phase Liquid Chromatography (RPLC)
Reversed-phase liquid chromatography, typically using octadecylsiloxane-bonded stationary phases and aqueous-organic mobile phases, is particularly effective for determining S, A, and B descriptors [30]. The high cohesive energy of water provides a strong driving force for solutes to interact with the stationary phase, while the addition of organic modifiers (acetonitrile, methanol) systematically changes the system constants, allowing multiple data points to be collected for each solute [30]. For compounds exhibiting variable hydrogen-bond basicity, the B° descriptor is typically used in RPLC systems [32].
Experimental Protocol for Descriptor Determination Using RPLC:
- Select a C18 or similar reversed-phase column
- Prepare mobile phases with varying proportions of water and organic modifier (e.g., 30-70% methanol or acetonitrile)
- Measure retention factors for the target compound and reference compounds with known descriptors
- Determine the system constants for each mobile phase composition using the reference compounds
- Use multiple retention measurements (at least 3-4 mobile phase compositions) to solve for the solute descriptors
- Apply statistical tools to identify and remove outliers or unreliable values
The main advantage of RPLC for descriptor determination is the ability to easily vary separation selectivity by changing mobile phase composition, providing multiple data points from a single column [30].
Micellar and Microemulsion Electrokinetic Chromatography (MEKC/MEEKC)
These electrokinetic techniques use micellar or microemulsion pseudostationary phases and are particularly useful for determining descriptors for ionic and ionizable compounds in addition to neutral molecules [30] [32]. The pseudostationary phases, typically composed of surfactants like sodium dodecyl sulfate, provide a distinct interaction environment complementary to GC and RPLC systems [30]. The determination process involves measuring retention factors at different pseudostationary phase concentrations and using the system constants derived from reference compounds with known descriptors [30].
Partitioning and Solubility Methods
Liquid-liquid partitioning methods between water and organic solvents or between two organic solvents provide direct measurement of partition coefficients (log P), which are used in the solvation parameter model for determining S, A, and B descriptors [30] [35]. The octanol-water system is the most widely characterized, but other systems like hexane-acetonitrile, chloroform-water, and alkane-solvent systems provide complementary information [30] [34].
Experimental Protocol for Octanol-Water Partition Coefficient (Kow) Measurement:
- Pre-saturate n-octan-1-ol and purified water by mutually saturating the solvents
- Dissolve the target compound in the pre-saturated octanol or water phase
- Combine the phases in a sealed container and equilibrate with constant agitation
- Separate the phases after equilibration (typically 24-48 hours)
- Analyze the concentration in one or both phases using appropriate analytical methods (GC, HPLC, etc.)
- Calculate log Kow = log(Coctanol/Cwater)
For the measurement of neutral PFAS descriptors, the shared-headspace and batch partition methods have been successfully employed, with verification through comparison to predictions from quantum chemically based models like COSMOtherm [34].
Totally organic biphasic systems, such as those containing triethylamine-formamide or ethanolamine-based systems, have been developed to extend the descriptor space for determining hydrogen-bond acidity and basicity descriptors [35]. These systems are particularly valuable for compounds with limited water solubility.
Optimized Descriptor Databases and Quality Control
The quality of solute descriptors depends critically on the reliability of the experimental data used in their determination. Two major curated databases exist: the Abraham compound descriptor database with over 8000 compounds, and the Wayne State University (WSU) compound descriptor database [32]. The WSU-2025 database represents an updated and expanded version containing optimized descriptors for 387 varied compounds, demonstrating improved precision and predictive capability compared to its predecessor [32].
The WSU database development employed rigorous quality control measures, including:
- Experimental data acquired in collaborating laboratories using consistent quality control and calibration protocols
- Application of screening tools to identify false experimental data associated with secondary compound-system interactions
- Use of the Solver method for simultaneous descriptor determination from multiple experimental measurements
- Statistical validation using theoretical models and comparison with experimentally determined values [32]
Table 2: Comparison of Major Solute Descriptor Databases
| Database | Number of Compounds | Data Sources | Quality Control Measures | Primary Applications |
|---|---|---|---|---|
| Abraham Database | >8000 | Combination of in-house measurements, literature data, property estimation methods | Variable quality due to diverse sources | Broad screening applications, general QSPR |
| WSU-2025 Database | 387 | Homogeneous experimental data from collaborating laboratories | Strict calibration protocols, statistical screening, Solver optimization | High-precision prediction, column characterization, environmental modeling |
The Solver method for descriptor determination involves using multiple (typically 5-8) experimental retention factors or partition coefficients measured in systems with known coefficients to establish an over-determined set of equations that can be solved for the solute descriptors [32]. This approach allows for the simultaneous determination of descriptors as a group, providing more robust values than single-technique approaches [30] [32].
Advanced Applications and Recent Developments
Case Study: PFAS Descriptor Determination
A recent application of these experimental techniques involved the comprehensive characterization of neutral per- and polyfluoroalkyl substances (PFAS) [34]. This study employed isothermal gas chromatography with four columns of differing polarity combined with octanol-water partition coefficient measurements to determine complete descriptor sets for 47 PFAS. The research revealed that PFAS with perfluoroalkyl chain lengths ≥4 show characteristic partition properties compared to non-PFAS, primarily due to the influence of the strongly electron-withdrawing perfluoroalkyl group on polar functional groups [34]. For instance, the hydrogen-bond acidity (A) of fluorotelomer alcohols was found to be higher than that of nonfluorinated alkyl alcohols, while the hydrogen-bond basicity (B) showed the opposite relationship [34].
Interconnection with Thermodynamic Models
Recent work has explored the interconnection between LSER databases and equation-of-state thermodynamics through the development of Partial Solvation Parameters (PSP) [1]. This approach aims to extract thermodynamic information from the LSER database for use in molecular thermodynamics, addressing the challenge of translating between different scales of intermolecular interactions [1]. The PSP framework includes two hydrogen-bonding parameters (σa and σb) reflecting acidity and basicity characteristics, a dispersion parameter (σd) for weak dispersive interactions, and a polar parameter (σp) for Keesom-type and Debye-type polar interactions [1].
Experimental Workflow and Research Tools
Diagram 1: Workflow for Experimental Determination of Solute Descriptors
Table 3: Essential Research Reagents and Materials for Descriptor Determination
| Reagent/Material | Specification | Primary Function | Application Techniques |
|---|---|---|---|
| n-Hexadecane | High purity (>99%) | Reference solvent for L descriptor definition | Gas chromatography, direct partitioning |
| Squalane | Chromatographic grade | Low-polarity stationary phase for GC | L descriptor determination |
| Poly(methyloctylsiloxane) | Immobilized stationary phase | Low-polarity GC phase with minimal hydrogen-bond basicity | L and S descriptor determination |
| C18-Bonded Silica | High purity, end-capped | Reversed-phase stationary phase | RPLC for S, A, B descriptor determination |
| n-Octan-1-ol | >99.5% purity | Organic phase for partition coefficients | Octanol-water partitioning |
| Sodium Dodecyl Sulfate | Electrophoresis purity | Surfactant for pseudostationary phases | MEKC/MEEKC |
| Reference Compounds | Varied structures with known descriptors | System calibration and method validation | All chromatographic and partitioning methods |
The experimental determination of solute descriptors through chromatographic and partitioning methods provides a robust foundation for applying the solvation parameter model across diverse scientific disciplines. The continued refinement of descriptor databases, particularly through homogeneous experimental data and rigorous quality control as demonstrated by the WSU-2025 database, enhances the precision and reliability of predictive models for chemical, biological, and environmental distribution processes. For researchers in drug development, these experimental techniques offer a efficient pathway to characterize molecular properties critical to understanding pharmacokinetic behavior without resorting to extensive in vivo testing. As methodological advancements continue, particularly in the integration of thermodynamic models and handling of challenging compound classes like PFAS, the utility of these experimental approaches will further expand, solidifying their role as essential tools in molecular property characterization.
Linear Solvation Energy Relationship (LSER) solute descriptors are a set of quantitatively defined parameters that encode key molecular properties influencing a solute's behavior in chemical and biological systems. The descriptors, often symbolized as Vx, E, S, A, B, and L, provide a powerful framework for predicting physicochemical properties and pharmacokinetic outcomes including solubility, permeability, and distribution. The Vx descriptor represents the characteristic molecular volume, which influences cavity formation in solvation processes. The E descriptor indicates excess molar refraction, capturing dispersion interactions. The S descriptor quantifies dipolarity/polarizability, reflecting a molecule's ability to engage in dipole-dipole interactions. The A and B descriptors represent hydrogen-bond acidity and basicity, respectively, crucial for predicting solvation in protic solvents. Finally, the L descriptor defines the gas-hexadecane partition coefficient at 25°C, characterizing hydrophobic interactions [36].
In pharmaceutical research and drug discovery, these descriptors have become indispensable for constructing quantitative structure-property relationship (QSPR) and quantitative structure-activity relationship (QSAR) models. They enable researchers to move beyond simple structural representations to a more nuanced understanding of how fundamental molecular interactions govern solubility, permeability through biological membranes, and overall drug-likeness. The ability to accurately predict these descriptors directly from molecular structure using computational methods represents a significant advancement over laborious experimental measurements, allowing for high-throughput screening of virtual compound libraries and rational drug design [37] [38].
Computational Methodologies for Descriptor Prediction
Quantum Chemical Calculations
Quantum chemical methods, particularly Density Functional Theory (DFT), provide a first-principles approach to calculating several LSER descriptors with high accuracy. DFT computations can directly yield electronic properties crucial for descriptor determination, including molecular orbital energies (HOMO-LUMO gap), dipole moments, and electrostatic potential surfaces [39].
The HOMO-LUMO gap (Egap) is particularly valuable as it serves as a quantum chemical property instrumental in different chemical research areas, as it correlates with molecular stability and reactivity [39]. DFT functionals such as B3LYP with appropriate basis sets (e.g., 6-31G*) are commonly employed to optimize molecular geometry and compute electronic properties. These calculations enable the prediction of S (dipolarity/polarizability) from computed dipole moments and polarizabilities, while hydrogen-bonding descriptors A and B can be derived from molecular electrostatic potentials and atomic charges [39].
For the Vx descriptor, DFT-optimized structures provide accurate molecular volumes through spatial integration of the electron density isosurface. The E descriptor (excess molar refraction) can be correlated with computed polarizabilities and refractive indices. Recent advances have incorporated machine learning to enhance the accuracy of DFT-predicted properties, with some models achieving mean absolute errors of 0.16 eV and 0.13 eV for HOMO and LUMO energies, respectively [39].
QSPR and Machine Learning Approaches
Quantitative Structure-Property Relationship (QSPR) modeling represents a powerful alternative to quantum chemical methods, particularly for high-throughput screening. QSPR models establish statistical relationships between structural descriptors and target properties using various regression and machine learning algorithms [39] [38].
Descriptor-based QSPR models utilize predefined molecular descriptors encoding structural information. Common descriptors include topological indices, electronic parameters, and thermodynamic properties. For instance, the Mordred package can generate over 1,600 two-dimensional molecular descriptors capturing different aspects of molecular structure [38]. Random Forest algorithms have demonstrated excellent performance with these descriptors, achieving coefficient of determination (R²) values of 0.88 for aqueous solubility prediction [38].
Signature molecular descriptors offer a particularly insightful approach, as they systematically codify atomic environments within a molecule. A Signature is defined as an extended valence description of atoms, capturing their connectivity to a predefined extent of branching (height). Recent research has utilized atomic Signatures to develop robust QSPR models with forward-stepping multilinear regression, achieving regression coefficients (r²) of 0.86 and predictability (q²) of 0.76 for properties like HOMO-LUMO gap [39].
Fingerprint-based methods, particularly circular fingerprints like Morgan fingerprints or Extended-Connectivity Fingerprints (ECFPs), provide a dynamic representation of molecular structure without predefined descriptors. These algorithms encode all possible molecular structure bonds by analyzing different fragments and hashing them into fixed-length bit strings [38]. While slightly less accurate than descriptor-based models for some endpoints (R² of 0.81 for solubility), fingerprint methods offer superior interpretability for investigating the impact of specific functional groups on target properties [38].
Integrated and Specialized Approaches
Advanced computational frameworks often integrate multiple methodologies to enhance predictive accuracy. Molecular Dynamics (MD) simulations provide detailed atomic-level insights into solvation processes, particularly for predicting the L descriptor (gas-hexadecane partition coefficient). MD simulations model the time evolution of molecular systems, capturing thermodynamic properties of the solvation process through analysis of molecular interactions between solute and solvent molecules [37].
For complex pharmacokinetic properties influenced by multiple LSER descriptors, machine learning ensembles have shown remarkable performance. Recent studies have curated large-scale databases containing over 24,000 bioactivity records to develop QSAR models for ABC transporter interactions using combinations of multiple machine learning algorithms and chemical descriptors [40]. These models demonstrated excellent performance with an average correct classification rate (CCR) of 0.764 for substrate binding models and 0.839 for inhibition models [40].
Table 1: Performance Comparison of Computational Methods for Property Prediction
| Method | Theoretical Basis | Applicable Descriptors | Performance Metrics | Computational Cost |
|---|---|---|---|---|
| DFT Calculations | Quantum Mechanics | S, A, B (from electronic properties) | High accuracy for electronic properties | High |
| QSPR with Molecular Descriptors | Statistical Regression | All descriptors | R² = 0.88 for solubility [38] | Medium |
| Signature-Based QSPR | Fragmental Analysis | All descriptors | r² = 0.86, q² = 0.76 for Egap [39] | Low |
| Fingerprint-Based ML | Machine Learning | All descriptors | R² = 0.81 for solubility [38] | Low |
| Molecular Dynamics | Statistical Mechanics | L, Vx, A, B | Detailed solvation thermodynamics | Very High |
Experimental Protocols and Validation Frameworks
QSPR Model Development Protocol
The development of robust QSPR models for predicting LSER descriptors follows a standardized workflow with critical validation steps:
Step 1: Data Curation and Preparation Collect experimental data for LSER descriptors from reliable sources such as published databases and literature. The dataset should encompass diverse chemical structures to ensure broad applicability. For aqueous solubility prediction, studies have successfully utilized curated collections of over 8,400 unique organic compounds from databases like Vermeire's, Boobier's, and Delaney's [38]. Structural standardization is essential, including normalization of tautomeric forms, removal of duplicates, and adjustment for ionization states.
Step 2: Molecular Descriptor Calculation and Selection Calculate molecular descriptors using computational packages such as Mordred, Dragon, or RDKit. Initial descriptor pools often contain hundreds to thousands of descriptors. Apply feature selection techniques including correlation filtering (e.g., threshold of 0.1), removal of low-variance descriptors, and elimination of highly correlated descriptors to reduce dimensionality. This process typically reduces descriptor sets from initial 1,613 to approximately 177 optimized descriptors [38].
Step 3: Model Training and Validation Split the dataset into training (~80%) and test sets (~20%). Apply machine learning algorithms including Random Forest, Support Vector Machines, or Multiple Linear Regression. Optimize hyperparameters using cross-validation techniques. For model validation, utilize both internal (cross-validation) and external validation with completely independent test sets. External validation with reliable experimental measurements not used in model development provides the most rigorous assessment of predictive performance [38].
Step 4: Model Interpretation and Applicability Domain Apply interpretation techniques such as SHAP (SHapley Additive exPlanations) analysis to identify the most influential structural features for each LSER descriptor. Define the applicability domain of the model to identify compounds for which predictions are reliable based on their structural similarity to the training set compounds [38].
Quantum Chemical Calculation Protocol
Step 1: Molecular Geometry Optimization Begin with initial 3D structure generation from SMILES strings or other chemical representations. Perform conformational analysis to identify the lowest energy conformation. Conduct geometry optimization using DFT methods with functionals such as B3LYP and basis sets like 6-31G*. Verify the absence of imaginary frequencies through frequency calculations to ensure true energy minima.
Step 2: Electronic Property Calculation Using the optimized geometry, compute electronic properties including molecular orbital energies (HOMO and LUMO), electrostatic potential maps, dipole moment, and polarizability. These calculations typically employ the same functional but may use larger basis sets with polarization and diffuse functions for improved accuracy.
Step 3: Descriptor Calculation Calculate LSER descriptors from the computed electronic properties. The S descriptor can be derived from the computed dipole moment and polarizability. Hydrogen-bonding descriptors A and B are calculated from molecular electrostatic potentials using approaches such as the COSMO-RS method. The Vx descriptor is obtained from the molecular volume computed from the optimized geometry [39].
Step 4: Validation with Experimental Data Validate computational results against available experimental measurements of LSER descriptors. For properties with limited experimental data, use predicted descriptors to calculate physicochemical properties (e.g., solubility, partition coefficients) with established LSER equations and compare with experimental values.
Computational Workflows and Signaling Pathways
The prediction of LSER descriptors from molecular structure follows systematic computational workflows that integrate multiple methodologies. Below is a detailed workflow using the DOT language, illustrating the logical relationships and decision points in the computational prediction process.
Computational Workflow for LSER Descriptor Prediction from Molecular Structure
Research Reagent Solutions and Essential Tools
Table 2: Essential Computational Tools for LSER Descriptor Prediction
| Tool/Category | Specific Examples | Function | Applicable Descriptors |
|---|---|---|---|
| Quantum Chemistry Software | Gaussian, GAMESS, ORCA, NWChem | Molecular geometry optimization and electronic property calculation | S, A, B (from first principles) |
| Molecular Descriptor Generators | Mordred, Dragon, RDKit, PaDEL | Calculation of topological, electronic, and constitutional descriptors | All descriptors via QSPR |
| Fingerprinting Tools | RDKit, OpenBabel, ChemAxon | Generation of structural fingerprints (ECFP, FCFP) for ML models | All descriptors via QSPR |
| Machine Learning Libraries | Scikit-learn, TensorFlow, PyTorch | Implementation of RF, SVM, NN algorithms for QSPR model development | All descriptors |
| Curated Experimental Data | Vermeire's Database, Boobier's Database, DrugBank | Experimental solubility, permeability, and descriptor values for model training/validation | All descriptors |
| Solvation Simulation Tools | GROMACS, AMBER, NAMD | Molecular dynamics simulations for solvation thermodynamics | L, Vx, A, B |
| Specialized Descriptor Tools | Signature descriptor implementations | Atomic Signature calculation for fragment-based QSPR | All descriptors [39] |
Computational approaches for predicting LSER solute descriptors from molecular structure have matured significantly, offering researchers powerful tools for high-throughput screening and rational molecular design. The integration of quantum chemical methods with modern machine learning techniques has created a robust framework for accurately estimating Vx, E, S, A, B, and L descriptors directly from structural information. These computational predictions enable the application of LSER models to vast virtual compound libraries, facilitating the identification of promising candidates with optimal physicochemical properties for pharmaceutical applications.
As the field advances, several emerging trends promise to enhance predictive capabilities further. The curation of larger, more diverse experimental datasets continues to improve model accuracy and applicability domains. Integration of multi-fidelity data, combining high-quality experimental measurements with rapid computational estimates, offers a pragmatic approach to balancing accuracy with throughput. Furthermore, the development of increasingly interpretable machine learning models helps bridge the gap between predictive performance and mechanistic understanding, allowing researchers to extract meaningful structure-property relationships from complex models. These advances collectively strengthen the role of computational descriptor prediction as an essential component of modern molecular design and optimization workflows.
Linear Solvation Energy Relationship (LSER) descriptors represent a powerful and theoretically grounded approach for quantifying molecular interactions in Quantitative Structure-Activity Relationship (QSAR) studies. The widely adopted Abraham LSER model utilizes a set of six fundamental molecular descriptors that capture distinct aspects of solute-solvent interactions: Vx (McGowan's characteristic volume), E (excess molar refraction), S (dipolarity/polarizability), A (hydrogen-bond acidity), B (hydrogen-bond basicity), and L (the gas-hexadecane partition coefficient) [41]. These descriptors provide a comprehensive framework for predicting a molecule's behavior in biological systems and its physicochemical properties, forming the basis for robust QSAR models that transcend simple structural correlations.
The fundamental LSER equations express free energy relationships for solute transfer between phases. For partition coefficient (KG) and solvation energy (KE) predictions, the LSER takes the form of two primary equations [41]: LogKG = -ΔG12/2.303RT = cg2 + eg2E1 + sg2S1 + ag2A1 + bg2B1 + lg2L1 LogKE = -ΔH12/2.303RT = ce2 + ee2E1 + se2S1 + ae2A1 + be2B1 + le2L1 In these equations, the uppercase letters represent solute-specific molecular descriptors, while the lowercase coefficients represent complementary solvent-phase-specific parameters. This mathematical formulation allows researchers to model complex biochemical interactions through multivariate linear regression techniques, providing predictive insights into biological activities and physicochemical properties critical for drug development and environmental risk assessment [41] [42].
Theoretical Foundation of LSER Descriptors
Molecular Interpretation of LSER Parameters
Each LSER descriptor quantifies a specific aspect of molecular interaction capability, providing a comprehensive picture of how a compound will behave in different environments [41]:
Vx (McGowan's Characteristic Volume): This descriptor represents the molecular volume and primarily reflects the energy cost of forming a cavity in the solvent to accommodate the solute molecule. It is calculated from molecular structure and is related to dispersion interactions.
E (Excess Molar Refraction): This parameter quantifies polarizability contributions from n- and π-electrons. It is derived from the refractive index and indicates a molecule's ability to engage in polarization interactions, particularly important for aromatic compounds and molecules with conjugated systems.
S (Dipolarity/Polarizability): This descriptor captures a molecule's ability to engage in dipole-dipole and dipole-induced dipole interactions. It represents the combined effect of molecular dipole moment and polarizability on solvation energy.
A (Hydrogen-Bond Acidity): This parameter quantifies a molecule's ability to donate hydrogen bonds, reflecting the strength of its interaction with hydrogen-bond acceptor sites in the environment or biological target.
B (Hydrogen-Bond Basicity): This descriptor measures a molecule's capacity to accept hydrogen bonds, indicating the strength of its interaction with hydrogen-bond donor groups.
L (Gas-Hexadecane Partition Coefficient): This descriptor represents the logarithm of the partition coefficient between the gas phase and n-hexadecane at 298 K, serving as a measure of dispersion forces and cavity formation energy in a non-polar solvent.
Thermodynamic Basis and Molecular Interactions
The LSER framework is grounded in solution thermodynamics, where the free energy change of solute transfer between phases is linearly related to molecular interaction parameters [41]. The model successfully decomposes the overall solvation energy into contributions from different interaction types, providing unprecedented insight into the fundamental forces driving molecular partitioning and biological activity. This theoretical foundation enables researchers to move beyond empirical correlations toward mechanistically interpretable QSAR models that offer predictive power across compound classes [41].
Recent advances have focused on addressing thermodynamic inconsistencies in traditional LSER applications, particularly for self-solvation of hydrogen-bonded compounds where solute and solvent become identical. Quantum chemical approaches, particularly COSMO-type calculations, are now enabling more thermodynamically consistent reformulations of LSER models that maintain predictive accuracy while improving theoretical robustness [41].
Experimental Protocols for LSER Descriptor Implementation
Determination of LSER Descriptors
Protocol 1: Experimental Determination of Descriptors
For researchers requiring experimentally derived LSER descriptors, the following protocol establishes a standardized approach [41]:
Solvent System Selection: Identify appropriate solvent systems that specifically probe the targeted molecular interactions. Common systems include:
- Hexadecane/water for determining L descriptor
- Cyclohexane/water for bulk dispersion interactions
- Alcohol/water systems for hydrogen-bonding parameters
Chromatographic Measurements:
- Utilize Reverse-Phase High-Performance Liquid Chromatography (RP-HPLC) with standardized stationary phases
- For IAM (Immobilized Artificial Membrane) chromatography, use IAM.PC.DD2 columns to simulate membrane interactions
- Maintain constant temperature (typically 25°C) throughout measurements
- Employ appropriate mobile phase buffers to control ionization states
Partition Coefficient Determination:
- Measure partition coefficients in multiple solvent systems to deconvolute individual interaction contributions
- For ionizable compounds, introduce the mean net charge per molecule (δ) as an additional parameter in amended LSER equations
- Validate measurements with standard compounds of known descriptor values
Data Regression Analysis:
- Apply multilinear regression to experimental partition coefficients across different solvent systems
- Solve for descriptor values that provide the best fit across all measured systems
- Verify internal consistency through statistical validation metrics
Protocol 2: Computational Estimation Using Group Contribution Methods
For rapid estimation of LSER descriptors, group contribution methods provide a practical alternative [7]:
Molecular Fragmentation:
- Deconstruct the target molecule into fundamental organic structures and functional groups
- Identify all relevant molecular fragments with established contribution values
Descriptor Calculation:
- Apply additive rules for Vx estimation based on molecular volume contributions
- Utilize established π* values for functional groups contributing to dipolarity/polarizability
- Apply βm values for hydrogen-bond accepting groups
- Apply αm values for hydrogen-bond donating groups
Validation and Adjustment:
- Compare calculated values with experimentally determined descriptors for structurally similar compounds
- Apply correction factors for intramolecular interactions or steric effects
- Verify that estimated values fall within reasonable ranges for the compound class
Quantum Chemical Calculation of LSER Descriptors
Protocol 3: QC-LSER Using COSMO-Type Calculations
Advanced quantum chemical approaches enable ab initio descriptor calculation [41]:
Molecular Structure Optimization:
- Perform geometry optimization using density functional theory (DFT) with appropriate basis sets
- Conduct conformational analysis to identify lowest energy conformers
- Verify optimization through frequency calculations (no imaginary frequencies)
COSMO Calculations:
- Execute COSMO (Conductor-like Screening Model) calculations to obtain sigma profiles
- Calculate distribution of molecular surface charges and potentials
- Derive interaction parameters from the sigma surface screening charge densities
Descriptor Determination:
- Calculate electrostatic interaction descriptors from molecular surface charge distributions
- Derive hydrogen-bonding free energies, enthalpies, and entropies from interaction energies
- Account for conformational changes upon solvation through ensemble averaging
Validation:
- Compare calculated descriptors with experimental values for training set compounds
- Adjust computational parameters to improve agreement with experimental data
- Establish uncertainty estimates for computationally derived descriptors
Table 1: LSER Descriptor Estimation Rules for Common Functional Groups [7]
| Functional Group | Vx/100 Contribution | π* Contribution | βm Contribution | αm Contribution |
|---|---|---|---|---|
| -CH3 | 0.25 | 0.00 | 0.00 | 0.00 |
| -CH2- | 0.20 | 0.00 | 0.00 | 0.00 |
| -OH | 0.08 | 0.25 | 0.45 | 0.33 |
| -COOH | 0.35 | 0.35 | 0.45 | 0.65 |
| -NH2 | 0.15 | 0.20 | 0.50 | 0.25 |
| -CHO | 0.25 | 0.35 | 0.45 | 0.00 |
| -C6H5 | 0.65 | 0.40 | 0.15 | 0.00 |
| -NO2 | 0.25 | 0.50 | 0.25 | 0.00 |
QSAR Model Development with LSER Descriptors
Data Preparation and Preprocessing
The foundation of any robust QSAR model lies in careful data preparation [43] [44]:
Training Set Selection:
- Curate a structurally diverse set of compounds with known biological activities or properties
- Ensure adequate representation across the chemical space of interest
- Include 20-50 compounds for initial model development, with expansion for more complex endpoints
Descriptor Matrix Preparation:
- Calculate or compile LSER descriptors (Vx, E, S, A, B, L) for all compounds
- Address missing data through estimation or strategic omission
- Standardize descriptor values to comparable scales to prevent dominance by large-value descriptors
Biological Activity Data:
- Utilize consistent, reproducible biological endpoint measurements
- Express activity as log-transformed values (e.g., IC50, EC50) for linear modeling
- Verify activity measurements through replicate experiments where possible
Model Construction and Validation
The construction of LSER-based QSAR models follows a systematic workflow [44]:
Diagram 1: QSAR Model Development Workflow
Variable Selection:
- Evaluate descriptor relevance through correlation analysis
- Identify and remove highly correlated descriptors to minimize multicollinearity
- Apply feature selection algorithms (e.g., stepwise regression, genetic algorithms) to identify optimal descriptor combinations
Model Construction Techniques:
- Multiple Linear Regression (MLR): Traditional approach for linear relationships between descriptors and activity [45]
- Partial Least Squares (PLS): Preferred method for handling descriptor collinearity [43] [45]
- Machine Learning Methods: Random Forest, Support Vector Machines, or Artificial Neural Networks for capturing non-linear relationships [44] [45]
Model Validation:
- Internal Validation: Apply leave-one-out or leave-multiple-out cross-validation to assess model robustness [43] [45]
- External Validation: Reserve a portion of compounds (20-30%) as an external test set never used in model development [44]
- Y-Scrambling: Randomize response variables to verify absence of chance correlations [43]
- Applicability Domain: Define chemical space where model predictions are reliable based on training set similarity [44]
Table 2: Statistical Measures for QSAR Model Validation [43] [44] [45]
| Validation Metric | Calculation | Acceptance Criterion | Purpose | ||
|---|---|---|---|---|---|
| R² (Training) | 1 - (SSres/SStot) | >0.6 | Goodness of fit for training set | ||
| Q² (LOO-CV) | 1 - (PRESS/SStot) | >0.5 | Internal predictive ability | ||
| R² (Test) | 1 - (SSres/SStot) | >0.6 | External predictive ability | ||
| RMSE | √(Σ(ŷi - yi)²/n) | Context-dependent | Prediction error magnitude | ||
| MAE | Σ | ŷi - yi | /n | Context-dependent | Average prediction error |
Advanced Integration Methodologies
Hybrid and Local QSAR Approaches
Modern QSAR methodologies have evolved beyond global models to incorporate localized and hybrid approaches:
aiQSAR Methodology [44]: The aiQSAR approach represents a significant advancement through runtime generation of local models specific to individual compounds:
Local Group Selection:
- Calculate structural fingerprints (PubChem and extended fingerprints) for all compounds
- Compute Tanimoto similarity distances between target compound and training set
- Select 20-50 most similar compounds to form the local modeling group
Descriptor Filtering:
- Remove descriptors with missing values for any compound in the local group
- Eliminate zero-variance and near-zero-variance descriptors
- Apply independent descriptor selection for each target compound
Multi-Model Consensus:
- Apply diverse modeling algorithms (glmboost, gaussprPoly, glmnet, rf, extratrees)
- Generate predictions from all methods
- Calculate consensus prediction through mean (regression) or majority vote (classification)
Applicability Domain Assessment:
- Compute Applicability Domain Measure (ADM) based on fingerprint similarities within local group
- Assign ADM rank (1-5) indicating prediction reliability
- Reject predictions below desired ADM threshold
Partial Order Ranking QSAR [46]: An alternative to conventional statistical methods that does not require assumptions of specific functional relationships:
Compound Ranking:
- Rank compounds based on descriptor values without assuming linear relationships
- Identify comparable compounds where all descriptors of one compound have equal or higher values
- Construct Hasse diagrams to visualize complex relationships
Prediction Uncertainty:
- Estimate uncertainty through distance between minAbove and maxBelow elements
- Refine model precision by increasing training set density in chemical space
3D-QSAR with LSER Descriptors
Three-dimensional QSAR approaches enhance predictive capability through spatial representation [43]:
Molecular Alignment:
- Superimpose training set compounds based on common pharmacophoric features
- Utilize crystallographic data or molecular superimposition software
- Ensure consistent orientation for field calculations
Interaction Field Calculation:
- Compute steric fields using Lennard-Jones potentials
- Calculate electrostatic fields using Coulombic potentials
- Incorporate LSER descriptors as additional constraints in field analysis
Partial Least Squares Regression:
- Apply PLS to correlate interaction fields with biological activity
- Generate coefficient contour maps visualizing important spatial regions
- Guide molecular optimization through visualization of favorable/unfavorable interactions
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for LSER-QSAR Studies
| Resource Category | Specific Tools/Resources | Function in LSER-QSAR |
|---|---|---|
| Computational Software | Dragon 7 [44] | Calculates 3839 molecular descriptors for comprehensive characterization |
| Quantum Chemistry Packages | COSMO-type calculators [41] | Derives LSER descriptors from first principles using quantum chemical calculations |
| Statistical Analysis | R packages: caret, pls, fingerprint [44] | Provides machine learning algorithms, PLS regression, and similarity calculations |
| QSAR Specialized Tools | lazar framework, T.E.S.T. [44] | Offers integrated QSAR modeling environments with automated workflows |
| Chemical Databases | PubChem [44] | Sources structural information and experimental bioactivity data |
| Chromatography Systems | IAM.PC.DD2 columns [42] | Determines membrane partitioning behavior for experimental descriptor validation |
| Descriptor Resources | LSER Database [41] | Provides compiled LSER descriptors and coefficients for diverse compounds |
Diagram 2: LSER Descriptor Generation Pathways
Applications in Drug Discovery and Development
The integration of LSER descriptors into QSAR modeling has enabled significant advances in pharmaceutical research:
Solubility and Permeability Prediction
LSER-based QSAR models excel at predicting critical physicochemical properties [46] [41]:
Aqueous Solubility Prediction:
- Apply LSER descriptors to model cavity formation energy in water
- Account for hydrogen-bonding contributions to solvation free energy
- Achieve correct ranking for 318 out of 319 compounds in validation studies [46]
Membrane Permeability:
- Utilize IAM chromatography to simulate membrane partitioning [42]
- Incorporate molecular electronic factors for ionizable compounds
- Predict absorption and distribution characteristics for drug candidates
Octanol-Water Partitioning:
- Model log P as fundamental hydrophobicity parameter
- Correctly rank 407 out of 408 compounds using partial order ranking methods [46]
- Establish reliable baseline for bioconcentration factor predictions
Toxicity and Environmental Risk Assessment
LSER descriptors provide mechanistic insight into toxicological endpoints:
Environmental Hazard Assessment:
- Predict bioaccumulation potential through LSER-based models
- Estimate toxicity toward specific organisms and cellular systems
- Support regulatory decisions through transparent, interpretable models [43]
Toxicity Prediction:
- Develop QSAR models for specific toxicological endpoints (e.g., skin corrosion, eye irritation)
- Apply TOPKIT and CaseTox software for specialized toxicity modeling [45]
- Group compounds with similar biological activity for read-across assessment
Future Perspectives and Methodological Advances
The continued evolution of LSER-QSAR integration faces both challenges and opportunities:
Quantum Chemical Enhancements
Emerging approaches leverage advanced computational chemistry to enhance LSER descriptors [41]:
COSMO-Based Descriptor Development:
- Derive molecular descriptors directly from quantum chemical calculations
- Establish thermodynamically consistent LSER equations
- Address self-solvation limitations of traditional LSER approaches
Conformational Dependence:
- Account for multiple molecular conformations in descriptor calculation
- Incorporate ensemble-averaged properties for flexible molecules
- Model conformational changes upon solvation or binding
Methodological Integration
Future developments will focus on hybrid approaches that combine strengths of multiple methodologies:
Multi-Model Consensus:
- Integrate predictions from local and global QSAR models
- Combine LSER descriptors with 3D-QSAR fields for comprehensive characterization
- Apply machine learning algorithms capable of handling mixed descriptor types
Automated Workflows:
- Develop streamlined pipelines for descriptor calculation and model validation
- Implement automated applicability domain assessment
- Create user-friendly interfaces for non-specialist researchers
The ongoing integration of LSER descriptors into QSAR modeling represents a powerful convergence of theoretical chemistry and practical predictive modeling, offering researchers an increasingly sophisticated toolkit for understanding and optimizing molecular properties across pharmaceutical, environmental, and materials science applications.
The blood-brain barrier (BBB) represents a formidable challenge in central nervous system (CNS) drug development, excluding over 98% of small-molecule drugs and nearly all large-molecule therapeutics from reaching the brain [47]. This comprehensive technical guide explores the application of Linear Solvation Energy Relationships (LSER) modeling to predict and enhance drug permeability across this protective barrier. By integrating the Abraham solvation parameter model with current BBB modulation strategies, we present a structured framework for researchers to quantify and predict solute-BBB interactions. The guide provides detailed methodologies for applying LSER principles, complemented by visualization of key pathways and tabular data for practical implementation in pre-clinical drug development workflows.
BBB Structure and Function
The blood-brain barrier is a highly selective semi-permeable membrane that separates circulating blood from the brain extracellular fluid in the central nervous system. This protective interface is primarily formed by brain microvascular endothelial cells connected by extensive tight junctions that severely limit paracellular diffusion [47] [48]. These endothelial cells are further supported by and interact with pericytes embedded within the vascular basement membrane, astrocytes whose end-feet encapsulate up to 99% of the endothelial surface, and various other cellular components that collectively constitute the neurovascular unit [47] [48].
From a drug delivery perspective, the BBB functions as a formidable gatekeeper that:
- Permits passive diffusion only for small (<400-600 Da), lipid-soluble molecules [47]
- Expresses active efflux transporters (e.g., P-glycoprotein) that pump compounds back into the bloodstream [47]
- Exhibits minimal transcellular vesicular transport (transcytosis) compared to peripheral endothelial [47]
- Maintains high transendothelial electrical resistance (TEER) through tight junction complexes [48]
The BBB Permeability Challenge in CNS Drug Development
The combined effects of these barrier properties create a significant bottleneck for neurological therapeutics. It has been estimated that the BBB excludes or limits the delivery of 98% of small-molecule drugs and nearly all large-molecule drugs to subtherapeutic levels [49] [47]. This limitation substantially complicates treatment strategies for CNS disorders including brain tumors, neurodegenerative diseases, and psychiatric conditions, often requiring innovative approaches to enhance therapeutic agent delivery across this protective interface.
LSER Fundamentals and Molecular Descriptors
Theoretical Basis of Linear Solvation Energy Relationships
The LSER model, also known as the Abraham solvation parameter model, provides a quantitative framework for correlating molecular structure and properties with thermodynamic parameters relevant to solute partitioning between different phases [12] [50] [1]. The model's power lies in its ability to deconstruct complex solute-solvent interactions into discrete, physically meaningful components that can be separately quantified and subsequently reassembled to predict partitioning behavior.
The fundamental LSER equations for processes involving partitioning between condensed phases and gas-to-solvent transfer are respectively expressed as:
log(P) = cp + epE + spS + apA + bpB + vpVx [1]
log(KS) = ck + ekE + skS + akA + bkB + lkL [1]
where P represents water-to-organic solvent partition coefficients, KS represents gas-to-organic solvent partition coefficients, the uppercase letters represent solute-specific molecular descriptors, and the lowercase coefficients represent system-specific complementary descriptors that characterize the solvent environment or biological barrier of interest.
Abraham Solute Descriptors and Their Physicochemical Significance
The LSER model utilizes six fundamental molecular descriptors that collectively capture the dominant interactions governing solute partitioning behavior:
Table 1: Abraham Solute Descriptors and Their Physicochemical Interpretation
| Descriptor | Symbol | Molecular Property Represented | Role in BBB Permeability |
|---|---|---|---|
| McGowan's Characteristic Volume | Vx | Molecular size and volume | Influences passive diffusion through lipid membranes |
| Excess Molar Refraction | E | Polarizability from n- and π-electrons | Affects van der Waals interactions with membrane components |
| Dipolarity/Polarizability | S | Dipole moment and molecular polarizability | Impacts interactions with polar membrane regions |
| Hydrogen Bond Acidity | A | Hydrogen bond donating ability | Reduces permeability through competition with membrane H-bond acceptors |
| Hydrogen Bond Basicity | B | Hydrogen bond accepting ability | Reduces permeability through competition with membrane H-bond donors |
| Gas-Hexadecane Partition Coefficient | L | Overall lipophilicity at molecular level | Primary driver for passive transcellular diffusion |
These descriptors provide a comprehensive framework for quantifying key molecular properties that influence a compound's ability to cross biological barriers, with particular relevance to the specific molecular interactions present at the BBB interface.
LSER Modeling for BBB Permeability Prediction
Adaptation of LSER Equations for BBB Permeability
For predicting blood-brain barrier permeability, the general LSER framework can be adapted to specifically model the partitioning of compounds between systemic circulation and brain tissue. The modified equation takes the form:
log(BBBP) = cBBB + eBBBE + sBBBS + aBBBA + bBBBB + vBBBVx + lBBBL
where BBBP represents the blood-brain barrier permeability measure (such as logBB or logPS), the uppercase variables remain the solute descriptors as defined in Table 1, and the lowercase coefficients with BBB subscript represent the system-specific parameters for the blood-brain barrier.
The system coefficients reflect the complementary properties of the BBB environment:
- vBBB: Reflects the barrier's responsiveness to solute volume (typically negative, indicating size exclusion)
- lBBB: Captures the barrier's sensitivity to solute lipophilicity (typically positive, favoring lipid-soluble compounds)
- aBBB and bBBB: Represent the barrier's hydrogen-bonding character (typically negative, indicating resistance to H-bonding compounds)
- eBBB and sBBB: Characterize the barrier's polar interactions with solutes
Experimental Protocols for BBB Permeability Determination
In Vivo Permeability Assessment
Materials Required:
- Animal model (typically mouse or rat)
- Test compounds at pharmacologically relevant concentrations
- Radiolabeled or fluorescent markers for quantification
- Surgical equipment for intravenous administration and sample collection
- Analytical instrumentation (HPLC-MS, scintillation counter, or fluorescence detector)
Procedure:
- Administer test compound intravenously to animals, ensuring precise dosing
- Allow sufficient circulation time for compound distribution (typically 5-60 minutes)
- Collect blood samples via cardiac puncture or other appropriate method
- Perfuse animals with saline to remove residual blood from cerebral vasculature
- Harvest brain tissue and homogenize in appropriate buffer
- Extract compound from brain homogenate and blood plasma using suitable techniques
- Quantify compound concentrations in both matrices using appropriate analytical methods
- Calculate permeability metrics such as logBB = log(Cbrain/Cblood) or more sophisticated measures like permeability-surface area product (PS)
In Vitro BBB Model Development
Materials Required:
- Brain endothelial cells (primary cultures or cell lines)
- Transwell culture plates with permeable membranes
- Cell culture media and supplements
- TEER measurement apparatus
- Permeability markers of various molecular weights
- Analytical instrumentation for compound quantification
Procedure:
- Culture brain endothelial cells on collagen-coated Transwell membranes
- Monitor transendothelial electrical resistance (TEER) regularly until values exceed 150-200 Ω·cm², indicating tight junction formation
- Validate barrier integrity using permeability markers such as sucrose, mannitol, or dextrans
- Apply test compounds to the donor (apical) compartment
- Sample from the receiver (basolateral) compartment at predetermined time points
- Quantify compound appearance in receiver compartment using HPLC-MS or other appropriate methods
- Calculate apparent permeability coefficients (P_app)
- Correlate in vitro P_app values with in vivo permeability data for model validation
Computational Implementation of BBB-LSER Models
Descriptor Calculation and Model Training
The successful application of LSER models for BBB permeability prediction requires careful computational implementation:
Descriptor Calculation: Compute Abraham descriptors for training set compounds using:
- Experimental measurement of partition coefficients in standardized systems
- Computational estimation using specialized software (e.g., ABSOLV, ACD/Percepta)
- Fragment-based contribution methods
Experimental Permeability Data Collection: Compile high-quality BBB permeability data from:
- In vivo measurements in rodent models
- In vitro permeability assays using validated BBB models
- Historical data from scientific literature (with careful attention to consistency)
Model Parameterization: Use multiple linear regression to determine the system-specific coefficients (cBBB, eBBB, sBBB, aBBB, bBBB, vBBB, lBBB) that best fit the experimental permeability data.
Model Validation: Employ rigorous cross-validation techniques and external test sets to evaluate predictive performance and domain of applicability.
The workflow below illustrates the integrated computational and experimental approach to developing LSER models for BBB permeability prediction:
Integration with Advanced BBB Modulation Strategies
Physical BBB Modulation Approaches
Recent advances in physical BBB modulation techniques offer complementary strategies for enhancing drug delivery to the CNS. LSER modeling can help identify compounds that would benefit most from these enhancement approaches:
Laser-Induced BBB Modulation
Gold Nanoparticle-Mediated Laser Stimulation:
- Mechanism: Tight junction-targeted gold nanoparticles (AuNP-BV11) activated by picosecond laser pulses (532 nm) cause temporary, reversible widening of tight junctions [49]
- Permeability Enhancement: Allows passage of immunoglobulins (∼150 kDa), viral vectors, and liposomes [49]
- Duration: Low laser fluence (≤5 mJ/cm²) causes BBB permeability for up to 6 hours; moderate fluence (10-25 mJ/cm²) for up to 72 hours [49]
- LSER Integration: Compounds with marginal permeability (moderately high A/B descriptors) may achieve therapeutic delivery with this approach
Low-Level Laser Treatment (LLLT):
- Mechanism: Infrared laser (1268 nm) generates singlet oxygen in endothelial cells and astrocytes without photosensitizers, temporarily decreasing tight junction protein expression [51]
- Application: Particularly valuable for pediatric GBM patients where traditional photodynamic therapy with photosensitizers is contraindicated [51]
- LSER Integration: Enables CNS delivery of compounds with suboptimal descriptor profiles through transient barrier modulation
The following diagram illustrates the key mechanisms of laser-induced BBB opening and their relationship to compound characteristics:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagents for BBB Permeability and LSER Modeling Research
| Reagent/Material | Function/Application | Experimental Context |
|---|---|---|
| Gold nanoparticles (AuNP-BV11) | Targets junctional adhesion molecule A (JAM-A) at tight junctions | Laser-induced BBB opening studies [49] |
| Transwell culture systems | Provides semi-permeable membrane support for in vitro BBB models | Barrier integrity assessment and permeability screening |
| TEER measurement equipment | Quantifies transendothelial electrical resistance as barrier integrity indicator | In vitro BBB model validation |
| Abraham descriptor calculation software (e.g., ABSOLV) | Computes molecular descriptors from chemical structure | LSER model implementation |
| Standardized solvent systems | n-hexadecane, octanol, water for experimental partition coefficient determination | LSER descriptor measurement |
| Radiolabeled or fluorescent tracers | (³H-sucrose, ¹⁴C-mannitol, FITC-dextrans) for permeability assessment | Barrier integrity validation |
| LC-MS/MS instrumentation | Sensitive quantification of test compounds in biological matrices | Permeability coefficient determination |
| Picosecond laser systems | (532 nm wavelength) for nanoparticle-mediated BBB modulation | Physical BBB opening methodologies |
Data Analysis and Interpretation Framework
Quantitative Analysis of BBB Permeability Using LSER
Successful application of LSER models requires careful interpretation of the system-specific coefficients derived for BBB permeability. The table below summarizes typical coefficient values and their interpretation:
Table 3: Interpretation of BBB-LSER System Coefficients
| System Coefficient | Typical Value Range | Physicochemical Interpretation | Implications for Drug Design |
|---|---|---|---|
| vBBB (Volume) | Negative (-0.5 to -1.5) | Steric hindrance and size exclusion | Favor compounds with molecular weight <500 Da |
| lBBB (Lipophilicity) | Positive (+0.5 to +1.5) | Favors partitioning into lipid membranes | Optimal log P ∼ 2-3 for CNS penetration |
| aBBB (H-bond Acidity) | Negative (-1.0 to -3.0) | Resistance to H-bond donors | Minimize hydrogen bond donors (<3) |
| bBBB (H-bond Basicity) | Negative (-2.0 to -4.0) | Resistance to H-bond acceptors | Minimize hydrogen bond acceptors (<7) |
| sBBB (Polarizability) | Variable (±0.5) | Dipolar interactions with membrane | Moderate effect compared to H-bonding |
| eBBB (Excess Refraction) | Variable (±0.3) | Polarizability from π-electrons | Limited impact on permeability |
Case Studies and Validation Approaches
To ensure model reliability, implement the following validation framework:
- Internal Validation: Use leave-one-out or k-fold cross-validation to assess predictive performance within the training set
- External Validation: Reserve a representative test set (20-30% of compounds) not used in model development
- Domain of Applicability: Define the chemical space boundaries where the model provides reliable predictions
- Experimental Corroboration: For critical compounds, validate predictions using in vitro BBB models or appropriate surrogates
The integration of LSER modeling with contemporary BBB research provides a powerful framework for rational CNS drug design. The quantitative structure-permeability relationships derived through the Abraham solvation parameter model enable researchers to prioritize compounds with favorable BBB penetration characteristics early in the development pipeline. Furthermore, the combination of LSER predictions with emerging physical BBB modulation technologies offers promising avenues for enhancing delivery of compounds that would otherwise be excluded from the brain.
Future developments in this field will likely focus on:
- Integration of LSER with machine learning approaches for improved prediction accuracy
- Development of cell-specific LSER models targeting different transport mechanisms at the BBB
- Application of LSER principles to predict drug delivery via novel BBB modulation techniques
- Expansion of descriptor databases to encompass increasingly diverse chemical space
As BBB penetration remains a critical determinant of CNS drug efficacy, the continued refinement and application of LSER modeling approaches will play an essential role in accelerating the development of therapeutics for neurological disorders.
Linear Solvation Energy Relationships (LSERs) represent a powerful thermodynamic framework for predicting key physicochemical properties in drug development. The widely adopted Abraham LSER model expresses a solute's free-energy related property, such as a partition coefficient, through the equation: SP = c + eE + sS + aA + bB + vV, where SP is any free-energy related property of a solute (e.g., log K) [52]. Each variable in this equation represents a specific molecular interaction descriptor: Vx (McGowan's characteristic volume), E (excess molar refraction), S (dipolarity/polarizability), A (hydrogen-bond acidity), B (hydrogen-bond basicity), and L (the gas-liquid partition coefficient in n-hexadecane at 298 K) [53]. These descriptors provide a quantitative basis for understanding how molecular structure influences solubility, permeability, and ultimately bioavailability.
In modern pharmaceutical development, the ability to accurately forecast solubility and permeability has become increasingly critical as an estimated more than 40% of new drug candidates are lipophilic and exhibit poor aqueous solubility [54]. This comprehensive technical guide examines current methodologies, computational approaches, and experimental protocols for applying LSER-based forecasting in preformulation studies and lead optimization, with particular emphasis on addressing the critical solubility-permeability interplay that dictates oral absorption success.
Fundamental Principles of LSER Modeling
Abraham LSER Descriptors: Molecular Interpretation
The Abraham LSER descriptors quantify specific aspects of molecular structure that govern solvation interactions. The McGowan's characteristic volume (Vx) represents the molar volume and relates to the energy required to form a cavity in the solvent [53]. The excess molar refraction (E) accounts for polarizability contributions from n- and π-electrons. The dipolarity/polarizability (S) descriptor characterizes a molecule's ability to engage in dipole-dipole and dipole-induced dipole interactions. The hydrogen-bonding descriptors (A and B) quantify hydrogen-bond donating (acidity) and accepting (basicity) capabilities, respectively, which are particularly crucial for pharmaceutical compounds as they determine interaction with biological membranes and aqueous environments [53]. Finally, the L descriptor represents the gas-liquid partition coefficient in n-hexadecane, serving as a measure of dispersion interactions and cavity formation energy in an inert solvent [53].
The thermodynamic basis of LSER models enables their application across multiple phases, with the general form adapting to specific partition processes. For solute transfer between gas and liquid phases, the equilibrium constant (KG) of solute partitioning is expressed as:
Similarly, for solvation energy (enthalpy) constant (KE):
These linear relationships provide direct connection to phase equilibrium calculations through the working equation:
where Vm2 is the molar volume of the solvent and γ1/2∞ is the activity coefficient of solute 1 at infinite dilution in solvent 2 [53].
Thermodynamic Foundation and Limitations
The LSER model fundamentally represents a three-step thermodynamic process that quantifies the free energy changes associated with solute transfer between phases [52]. While exceptionally valuable, traditional LSER approaches face two primary limitations: their expansion is restricted by the availability of experimental data for regression analysis, and they can demonstrate thermodynamic inconsistency when applied to self-solvation of hydrogen-bonded solutes [53]. This inconsistency manifests particularly when solute and solvent become identical in self-solvation situations, where the expected equality of complementary hydrogen-bonding interaction energies is not maintained [53].
Recent advances address these limitations through quantum chemical calculations that enable thermodynamically consistent reformulation of QSPR-type Linear Free-Energy Relationship models [53]. These approaches derive new molecular descriptors of electrostatic interactions from the distribution of molecular surface charges obtained from COSMO-type quantum chemical calculations, allowing more robust prediction of hydrogen-bonding free energies, enthalpies, and entropies for diverse solutes [53].
Table 1: Abraham LSER Descriptors and Their Molecular Significance
| Descriptor | Molecular Interpretation | Primary Role in Solubility/Permeability |
|---|---|---|
| Vx | McGowan's characteristic volume | Determines cavity formation energy in solvent |
| E | Excess molar refraction | Quantifies polarizability from n- and π-electrons |
| S | Dipolarity/polarizability | Characterizes dipole-dipole and induced dipole interactions |
| A | Hydrogen-bond acidity | Measures hydrogen-bond donating capacity |
| B | Hydrogen-bond basicity | Measures hydrogen-bond accepting capacity |
| L | n-Hexadecane/air partition coefficient | Represents dispersion interactions and cavity formation |
Computational Approaches for Solubility Prediction
Machine Learning Models for Solubility Forecasting
Recent advances in machine learning have significantly improved computational solubility prediction. Researchers at MIT have developed models that demonstrate two to three times greater accuracy compared to previous approaches like the Abraham Solvation Model or SolProp [55]. Two primary architectures have shown particular promise: FastProp, which incorporates static molecular embeddings, and ChemProp, which learns molecular embeddings during training [55]. Surprisingly, despite their different approaches, both models perform essentially equally well when trained on comprehensive datasets like BigSolDB, suggesting that data quality rather than model architecture currently limits prediction accuracy [55].
These models excel at predicting temperature-dependent solubility variations, a crucial advantage for pharmaceutical processing. Their performance stems from training on extensive compiled datasets, with BigSolDB containing approximately 40,000 data points from nearly 800 published papers, covering about 800 molecules dissolved in more than 100 organic solvents [55]. The models have proven particularly valuable for identifying less hazardous alternative solvents that minimize environmental and physiological damage while maintaining sufficient solvation capacity [55].
Quantum Chemical Calculations and COSMO-RS
The integration of quantum chemical calculations with LSER methodologies represents a significant advancement in solubility prediction. COSMO-RS (Conductor-like Screening Model for Real Solvents) adopts a simple nearest-neighbor pairwise additive interaction approach combined with detailed quantum-chemical information on molecular charge density distributions [53]. This combination creates a powerful a priori predictive tool in Molecular Thermodynamics, particularly for solute solvation and partitioning calculations [53].
A key development is the derivation of new molecular descriptors based on molecular charge-density distributions or sigma-profiles from COSMO-RS calculations [53]. These descriptors enable the development of thermodynamically consistent linear solvation energy relationships that can effectively handle self-solvation scenarios and conformational changes during solvation [53]. While COSMO-RS cannot separately calculate hydrogen-bonding contributions to solvation free energy, it can determine the corresponding contribution to solvation enthalpy, allowing comparison with LSER contributions [53].
Table 2: Computational Models for Solubility Prediction
| Model | Methodology | Applications | Advantages |
|---|---|---|---|
| Abraham LSER | Linear free-energy relationships with experimentally derived descriptors | Solvent screening, partition coefficient prediction | Thermodynamic basis, interpretability |
| COSMO-RS | Quantum chemical calculations with pairwise surface segment interactions | A priori solubility prediction, hydrogen-bonding energy calculation | Less dependent on experimental data, handles novel structures |
| FastSolv/FastProp | Machine learning with static molecular embeddings | High-throughput solubility screening, solvent selection | Fast predictions, high accuracy for known chemical space |
| ChemProp | Message-passing neural networks with learned embeddings | Novel molecule solubility prediction, temperature-dependent solubility | Adapts to new structural patterns, strong extrapolation capability |
| Ensemble ML Models | Combined XGBR, LGBR, and CATr with bio-inspired optimization | Supercritical CO₂ systems, complex thermodynamic conditions | Handles strong non-linearities, high fidelity for specialized applications |
Advanced Ensemble Approaches for Complex Systems
For particularly challenging prediction scenarios such as drug solubility in supercritical CO₂ (SCCO₂) systems, advanced ensemble machine learning frameworks have demonstrated remarkable efficacy. Recent research combines Extreme Gradient Boosting Regression (XGBR), Light Gradient Boosting Regression (LGBR), and CatBoost Regression (CATr), facilitated by bio-inspired optimization algorithms like the Artificial Protozoa Optimizer (APO) and Hippopotamus Optimization Algorithm (HOA) [56]. These ensembles achieve exceptional predictive accuracy (R² = 0.9920, RMSE = 0.08878) for pharmaceutical solubility in SCCO₂ by effectively capturing complex non-linear behaviors across varying thermodynamic conditions [56].
The robustness of these models is ensured through k-fold cross-validation, with interpretability enhanced via SHAP and FAST sensitivity analysis. The generation of prediction intervals using bootstrapping further enhances reliability for real-world pharmaceutical applications, providing confidence estimates for solubility predictions under specific temperature and pressure conditions [56].
Permeability Forecasting and the Solubility-Permeability Interplay
Fundamental Relationship Between Solubility and Permeability
The critical relationship between solubility and permeability represents one of the most significant considerations in oral drug development. Mathematical representation of intestinal permeability (Peff) includes the relationship: Peff = (D × K)/h, where D is the diffusion coefficient through the membrane, K is the membrane/aqueous partition coefficient, and h is the membrane thickness [54]. This direct correlation between intestinal permeability and membrane/aqueous partitioning, which in turn depends on the drug's apparent solubility in the GI milieu, establishes an inherent solubility-permeability interplay that must be considered in formulation development [54].
This interplay frequently manifests as a trade-off wherein formulation approaches that increase apparent solubility may decrease apparent permeability. For instance, when using cyclodextrin-based solubility-enabling formulations, the solubility increase is accompanied by a decrease in the drug's free fraction available for membrane permeability, potentially leading to paradoxical effects on overall absorption [54]. Understanding and quantifying this balance is essential for maximizing the overall fraction of drug absorbed.
Mass Transport Modeling for Permeability Prediction
Quantitative mass transport models have been developed to elucidate the impact of various formulation approaches on intestinal permeability. These models consider both intestinal membrane permeability (Pm) and unstirred water layer (UWL) permeability (Paq) to predict the overall effective permeability (Peff) dependence on formulation components [54]. For cyclodextrin-based systems, modeling reveals that: (1) UWL permeability increases with increasing cyclodextrin concentration due to decreased effective UWL thickness; (2) permeability through the intestinal membrane decreases with increasing cyclodextrin concentration, attributed to decreased free drug fraction; and (3) above certain cyclodextrin concentrations, the UWL is effectively abolished and overall Peff tends toward membrane control (Peff ≈ Pm) [54].
These models enable excellent quantitative prediction of permeability as a function of cyclodextrin concentrations across various permeability models, including PAMPA assays, Caco-2 studies, and in situ rat jejunal perfusion models [54]. The models demonstrate that overall drug absorption is governed by the tradeoff between solubility increase and permeability decrease, emphasizing the necessity to consider both parameters simultaneously during formulation development.
Diagram 1: The solubility-permeability interplay demonstrates how formulation strategies to enhance solubility often reduce permeability, requiring optimal balance for maximal absorption.
Experimental Protocols and Methodologies
LSER Determination for Custom-Made Phases
The application of LSERs to characterize custom-made phases follows a well-established experimental protocol. For solid-phase microextraction fibers, the methodology involves experimentally determining the log K for a series of solutes with known solute descriptors (E, S, A, B, and V) and performing multi-linear regression to obtain the unknown system coefficients (e, s, a, b, and v) [52]. The sign and magnitude of these system coefficients reflect the relative strengths of chemical interactions affecting partitioning between the two phases (fiber and water) [52]. Studies applying this methodology to custom-made polyaniline (PANI) fibers have demonstrated that the system properties having the greatest influence on log K were ease of cavity formation and hydrogen bond donating ability, with differences in dipolarity/polarizability and hydrogen bond accepting ability revealing unique partitioning environments across different fibers [52].
The experimental workflow consists of: (1) selecting a diverse set of probe molecules with known Abraham descriptors; (2) measuring partition coefficients for these probes between the custom phase and water; (3) performing multilinear regression to determine system-specific coefficients; (4) validating the model with test compounds; and (5) applying the characterized system to predict partitioning behavior for new compounds.
Solubility Measurement Protocols
Accurate solubility measurement is fundamental to LSER development and validation. Two method-dependent terms are used in pharmaceutical literature: kinetic solubility, defined as the concentration of solute in solvent when an induced precipitate first appears in solution, and thermodynamic solubility, defined as the concentration of compounds in solution when the solution is in equilibrium with solute in the presence of excess undissolved solute [57]. Thermodynamic solubility is considered the gold standard for optimizing poorly soluble lead compounds and depends on various factors including pH, temperature, ionic strength, salt/buffer effects, and phase separation [57].
The basic experimental distinction between these approaches lies in sample preparation: for thermodynamic solubility, solid-form compound is added to aqueous medium, while for kinetic solubility, pre-dissolved compound is used for determination [57]. Modern high-throughput methods for determining thermodynamic solubility include solid-state characterization by polarized light microscopy, Raman spectroscopy, powder X-ray diffraction, ultra-performance liquid chromatography, and polychromatic turbidimetry [57].
Structural Modification Strategies for Solubility Optimization
Structural modification represents a versatile medicinal chemistry approach for improving solubility while potentially optimizing other pharmacokinetic parameters simultaneously. Successful strategies include:
- Prodrug design: Incorporating cleavable hydrophilic groups that enhance aqueous solubility while maintaining parent compound activity after in vivo conversion [57].
- Hydrophilic and ionizable group insertion: Adding groups such as amines, carboxylic acids, or polyethylene glycol chains to increase water interaction capacity [57].
- Hydrogen bonding manipulation: Strategic addition or removal of hydrogen bond donors and acceptors to optimize crystal packing energy and solvation [57].
- Bioisosteric replacement: Substituting atoms or groups with alternatives that maintain similar biological activity but improve solubility profiles [57].
- Molecular symmetry and planarity disruption: Reducing molecular symmetry and planarity to decrease crystal lattice energy and improve dissolution [57].
These structural modifications target the fundamental factors affecting solubility, particularly lipophilicity (logP) and crystal lattice energy (represented by melting point), as captured in the general solubility equation: logSw = 0.5 - logP - 0.01(MP-25), where logP represents lipophilicity and MP represents melting point as an indicator of crystal lattice energy [57].
Table 3: Experimental Methods for Solubility and Permeability Assessment
| Method | Application | Key Parameters | Considerations |
|---|---|---|---|
| Thermodynamic Solubility | Gold standard for solubility optimization | Equilibrium concentration with excess solid | Requires careful control of pH, temperature, ionic strength |
| Kinetic Solubility | High-throughput screening in discovery | Concentration at precipitation onset | Uses DMSO stock solutions; may overestimate thermodynamic solubility |
| PAMPA | Passive membrane permeability prediction | Effective permeability across artificial membrane | Limited to passive diffusion mechanisms |
| Caco-2 Model | Intestinal permeability assessment | Apparent permeability and efflux ratios | Includes transporter effects; longer culture time required |
| In Situ Perfusion | Regional intestinal permeability in animals | Effective permeability in physiological environment | Resource-intensive; provides most physiologically relevant data |
| LSER Characterization | Phase partitioning behavior | System coefficients (e, s, a, b, v) | Requires multiple probe molecules with known descriptors |
Applications in Preformulation and Lead Optimization
Biopharmaceutical Classification System (BCS) Framework
The Biopharmaceutics Classification System (BCS) provides a fundamental framework for applying solubility and permeability forecasting in drug development. BCS classifies compounds into four categories based on solubility and permeability characteristics [54]. Molecules with poor solubility pose the greatest risk of low oral bioavailability, particularly those belonging to BCS Class II (lower solubility and higher permeability) and BCS Class IV (lower solubility and lower permeability) that require modification for solubility improvement [57]. Accurate classification early in development guides formulation strategy and identifies candidates requiring special intervention.
The role of solubility and permeability as key parameters controlling oral drug absorption makes their accurate prediction crucial throughout the drug discovery pipeline, from lead optimization to formulation development. The BCS framework enables scientists to anticipate absorption challenges and prioritize compounds with optimal solubility-permeability balance.
Integrating Computational and Experimental Approaches
Successful preformulation strategies integrate computational predictions with experimental validation. Computational tools provide early assessment of potential solubility issues, allowing medicinal chemists to implement structural modifications before extensive synthesis. The integration of machine learning models like FastSolv with traditional LSER approaches creates a powerful workflow for solvent selection in synthetic route development [55]. These integrated approaches are particularly valuable for identifying less hazardous alternative solvents that minimize environmental and physiological damage while maintaining sufficient solvation capacity, addressing the pharmaceutical industry's need for greener processes [55].
In lead optimization, forecasting models guide structural modifications to improve solubility without compromising permeability or biological activity. The solubility-permeability interplay must be carefully considered during these modifications, as changes that improve solubility may adversely affect permeability, potentially negating the overall absorption benefit [54]. Successful optimization requires balancing multiple parameters to achieve the optimal combination of solubility, permeability, and potency.
Table 4: Essential Research Tools for Solubility and Permeability Forecasting
| Resource | Category | Key Function | Application Context |
|---|---|---|---|
| Abraham LSER Database | Database | Comprehensive thermodynamic data for LSER calculations | Solvent system characterization, partition coefficient prediction |
| BigSolDB | Database | Curated solubility data for ~800 molecules in 100+ solvents | Machine learning model training and validation |
| COSMO-RS | Software | Quantum chemical calculations of solvation properties | A priori solubility prediction for novel compounds |
| FastSolv/FastProp | Software | Machine learning solubility prediction with static embeddings | High-throughput solvent screening for synthesis |
| ChemProp | Software | Message-passing neural networks for property prediction | Novel chemical space exploration with limited data |
| PAMPA | Assay System | Parallel artificial membrane permeability assay | Early-stage passive permeability screening |
| Caco-2 Model | Cell Culture | Human colorectal adenocarcinoma cell line | Intestinal permeability with transporter effects |
| Thermodynamic Solubility Assay | Experimental Protocol | Equilibrium solubility measurement under controlled conditions | Preformulation studies, BCS classification |
Structural Modification Toolkit for Medicinal Chemists
Medicinal chemists employ specific structural modifications to optimize solubility during lead optimization:
- Ionizable group incorporation: Adding basic amines or acidic carboxylic acids to enhance pH-dependent solubility, particularly for ionizable compounds where apparent solubility relates to intrinsic solubility through SpH = Si(1 + 10^(pH-pKa)δi), where δi = {1, -1} for acid and basic groups, respectively [57].
- Hydrogen bond optimization: Balancing hydrogen bond donors and acceptors to maintain target engagement while improving solvation energy.
- Lipophilicity reduction: Decreasing logP through introduction of polar groups or reduction of hydrophobic surface area.
- Crystal lattice energy disruption: Incorporating structural features that disrupt efficient crystal packing to reduce melting point and improve dissolution.
- Prodrug approaches: Designing reversible derivatives with enhanced solubility characteristics that convert to active parent compound in vivo.
These structural modifications require careful optimization since excessive hydrophilicity can compromise membrane permeability, demonstrating the critical solubility-permeability balance that must be maintained throughout lead optimization [54].
Diagram 2: Integrated workflow for lead optimization combining computational prediction and experimental validation in solubility-permeability optimization.
The integration of LSER frameworks with advanced computational approaches continues to evolve, offering increasingly accurate prediction of solubility and permeability characteristics critical to pharmaceutical development. Recent advances in quantum chemical calculations, machine learning models, and thermodynamically consistent LSER reformulations address longstanding limitations of traditional methods while maintaining interpretability [53] [55]. The recognition of the essential solubility-permeability interplay has fundamentally changed formulation strategies, emphasizing balanced optimization rather than unilateral solubility enhancement [54].
Future developments will likely focus on improved integration of first-principles calculations with machine learning, expanded databases for training models, and dynamic prediction frameworks that account for physiological changes throughout the gastrointestinal tract. As these forecasting capabilities mature, they will continue to reduce reliance on trial-and-error approaches, accelerating the development of bioavailable pharmaceuticals with optimal therapeutic profiles.
The Linear Solvation Energy Relationship (LSER) framework is a powerful quantitative approach for predicting the physicochemical properties and biological activities of organic compounds. In the context of percutaneous absorption, LSER models describe skin permeability as a function of a solute's intrinsic molecular descriptors, which represent its capability for different types of intermolecular interactions [58]. These models have gained prominence as reliable tools for predicting skin permeation, crucial for transdermal drug delivery development and chemical risk assessment [59] [60].
The standard LSER equation for skin permeability incorporates key solute descriptors that capture the dominant molecular interactions governing permeation through the stratum corneum, the skin's primary barrier layer [59]. This case study explores the application of these descriptors through the analysis of published experimental data, computational models, and practical implementation protocols for pharmaceutical researchers.
Theoretical Framework of LSER Descriptors
The LSER Equation for Skin Permeability
The permeation of a molecule through human skin can be described using the following LSER equation:
log Kp = c + vVx + eE + sS + aA + bB + lL
Where:
- Kp = Permeability coefficient (cm/h)
- Vx = McGowan's characteristic molecular volume in cm³/100
- E = Excess molar refractivity
- S = Dipolarity/polarizability
- A = Overall hydrogen-bond acidity
- B = Overall hydrogen-bond basicity
- L = Logarithm of hexadecane-air partition coefficient [58]
The coefficients (v, e, s, a, b, l) in the equation are regression coefficients that reflect the complementary properties of the skin membrane, while the solute descriptors (Vx, E, S, A, B, L) characterize the molecule's solvation properties.
Molecular Interactions Captured by LSER Descriptors
Table: LSER Solute Descriptors and Their Molecular Significance
| Descriptor | Symbol | Molecular Interpretation | Role in Skin Permeation |
|---|---|---|---|
| McGowan Volume | Vx | Molecular size/bulk | Quantifies steric effects and diffusion limitations through lipid bilayers |
| Excess Molar Refractivity | E | Electron lone pair interactions and polarizability | Captures dispersion forces with electron-rich membrane components |
| Dipolarity/Polarizability | S | Dipole-dipole and induced dipole interactions | Reflects interactions with polar head groups in stratum corneum lipids |
| Hydrogen-Bond Acidity | A | Hydrogen bond donating ability | Determines solvation with acceptor groups in skin proteins and lipids |
| Hydrogen-Bond Basicity | B | Hydrogen bond accepting ability | Governs interactions with donor groups in the skin barrier |
| Hexadecane-Air Partition | L | General hydrophobicity/lipophilicity | Predicts partitioning into lipophilic regions of stratum corneum |
The hydrogen-bonding parameters (A and B) are particularly critical for skin permeation prediction, as hydrogen bonding significantly influences solute partitioning between aqueous environments and the lipophilic stratum corneum [58]. Research has demonstrated that hydrogen bond strength and directionality are essential factors governing permeability coefficients [58].
Computational Implementation and Data Analysis
Experimental Skin Permeability Datasets
Recent advances in skin permeability prediction have been facilitated by the development of comprehensive, curated databases. The following table summarizes key datasets used in developing and validating LSER models:
Table: Experimental Skin Permeability Databases for LSER Modeling
| Database | Size | Data Content | Key Experimental Parameters | Access |
|---|---|---|---|---|
| HuskinDB | 253 substances | logKp, steady-state flux (Jss), maximum flux (Jmax), lag time (tlag) | Skin source (abdomen, breast, thigh), skin type (epidermis, dermis), donor concentration, temperature [61] [62] | Freely accessible online |
| SkinPiX | 441 records for 140 molecules | logKp, Jss, Jmax, tlag | Donor/receptor pH, skin integrity tests, vehicle composition, membrane type [61] [60] | Open-source dataset |
| FDA-Approved Drug Set | 2326 compounds | Predicted logKp values, molecular descriptors | Anatomical Therapeutic Chemical (ATC) classification, cluster analysis [60] | Derived from public sources |
These datasets highlight the importance of standardizing experimental conditions when building predictive models. Critical parameters include skin source (abdomen preferred), skin layer (epidermis), donor concentration (dilute vs. saturated), temperature (31-35°C to mimic skin surface), and pH (7-7.5 to represent physiological conditions) [61] [62].
LSER Model Performance and Validation
Modern implementation of LSER models frequently incorporates machine learning algorithms to capture nonlinear relationships in complex datasets [60]. Recent studies have demonstrated that ensemble methods like Light Gradient Boosting Machine (LGBM), XGBoost, and Random Forest outperform traditional multiple linear regression for predicting logKp values [60].
The predictive performance of these models is typically evaluated using:
- Coefficient of determination (R²): Values >0.7 indicate good predictive ability
- Root Mean Square Error (RMSE): Lower values indicate higher precision
- Cross-validation: Ensuring model robustness across different compound classes
For example, a recently developed fragment contribution model based on HuskinDB data achieved R² = 0.7125 and RMSE = 0.71 for the training set (n=29), and R² = 0.8931 with RMSE = 0.49 for the test set (n=7) [62].
Experimental Protocols for Skin Permeation Studies
In Vitro Diffusion Cell Methodology
The Franz-type diffusion cell remains the gold standard for experimental determination of skin permeability parameters [59] [61]. The following workflow details the protocol for generating high-quality data suitable for LSER modeling:
Diagram: Experimental Workflow for Skin Permeation Studies
Skin Membrane Preparation
- Source: Human abdominal skin is preferred; surgical discard or ethically sourced
- Storage: Frozen at -20°C until use (within 6 months maximum)
- Preparation: Heat separation (60°C for 45-60s) to isolate epidermis from dermis
- Thickness: 200-800 μm; measure with micrometer for consistency [61]
Diffusion Cell Assembly
- Cell Type: Static Franz-type diffusion cells with standard configuration
- Surface Area: Typically 0.5-1.0 cm² for research applications
- Receptor Volume: 3-5 mL to maintain sink conditions
- Receptor Fluid: Phosphate buffered saline (pH 7.4) for unionized solutes
- Temperature: Maintain at 32±1°C using circulating water jacket [61]
Compound Application and Sampling
- Dosing: Infinite dose conditions (saturated solution) for Kp determination
- Vehicle: Aqueous buffer (pH 7.0-7.5) preferred for standardization
- Sampling Intervals: 1, 2, 4, 6, 8, 12, 24, 36, 48 hours post-application
- Sample Volume: 200-500 μL with receptor fluid replacement [61]
Permeability Parameter Calculation
From the experimental data, three key parameters are derived:
Steady-state Flux (Jss): Calculated from the slope of the cumulative amount permeated versus time plot during the linear phase (μg/cm²/h)
Permeability Coefficient (Kp): Determined using the equation Kp = Jss/Cv, where Cv is the vehicle concentration (cm/h)
Lag Time (tlag): Obtained from the x-intercept of the linear portion of the permeation curve (h) [61]
These parameters form the foundation for developing and validating LSER models for skin permeability prediction.
Research Toolkit for LSER-Based Permeability Prediction
Table: Essential Research Reagents and Computational Tools
| Category | Item/Solution | Function/Application | Technical Specifications |
|---|---|---|---|
| Experimental Materials | Human epidermal membrane | Barrier for permeation studies | Abdomen source, 200-800 μm thickness [61] |
| Franz diffusion cells | Permeation experimental apparatus | Standard configuration, 0.5-1.0 cm² diffusion area [59] | |
| Phosphate buffered saline | Receptor fluid medium | pH 7.4, isotonic, maintained at 32°C [61] | |
| Test compounds in vehicle | Permeants for study | Aqueous solutions, unionized fraction >0.9 [60] | |
| Computational Tools | Chemistry Development Kit (CDK) | Molecular descriptor calculation | Open-source, calculates 1D/2D descriptors from SMILES [60] |
| Scikit-Learn | Machine learning implementation | Python library for regression models (RF, XGBoost, etc.) [60] | |
| R with ggplot2 | Statistical analysis and visualization | Open-source environment for LSER model development [63] | |
| Data Resources | HuskinDB | Human skin permeability database | 253 compounds with experimental parameters [62] |
| SkinPiX | Recent permeability data compilation | 441 records from 2012-2021 literature [61] |
Advanced Modeling Approaches
Integration of LSER with Machine Learning
Contemporary research has demonstrated that combining LSER descriptors with nonlinear machine learning algorithms significantly enhances prediction accuracy for skin permeability [60]. The optimal workflow involves:
- Descriptor Calculation: Computing LSER parameters from molecular structure
- Feature Selection: Identifying the most relevant descriptors (typically Vx, A, B, L)
- Model Training: Implementing ensemble methods like Random Forest or Gradient Boosting
- Validation: Assessing predictive performance on external test sets
This hybrid approach has shown particular utility for analyzing FDA-approved drugs, where cluster analysis based on LSER descriptors reveals distinct permeability patterns across different therapeutic classes [60].
Fragment Contribution Models as LSER Alternatives
For compounds with limited experimental data, fragment contribution models provide a complementary approach to traditional LSER methodology. These models predict permeability based solely on the presence and frequency of functional groups within a molecule [62].
The general form of a fragment contribution model is:
log Kp = Intercept + Σ(fragment coefficient × number of occurrences)
For example, the presence of aromatic rings contributes +0.168 to logKp, while carboxylic acid groups contribute -1.521, reflecting their opposing effects on permeability [62]. These simplified models demonstrate how LSER principles can be adapted for rapid screening in early drug development.
The application of LSER descriptors to predict skin permeation represents a robust, mechanistically grounded approach that continues to evolve with advances in computational chemistry and machine learning. By integrating traditional LSER methodology with modern data science techniques, researchers can develop increasingly accurate models to guide transdermal drug delivery system design and chemical safety assessment. The standardized experimental protocols and computational tools outlined in this case study provide a foundation for implementing these approaches in pharmaceutical research and development.
Linear Solvation Energy Relationships (LSERs) represent a cornerstone methodology in physical chemistry and pharmaceutical research for predicting and interpreting the partitioning behavior of solutes in different chemical environments. The most widely accepted model, known as the Abraham solvation parameter model, provides a robust framework for understanding solute-solvent interactions. For researchers, particularly in drug development, mastering the software and tools for LSER calculation is paramount for applications ranging from predicting drug solubility and permeability to optimizing chromatographic separations and assessing environmental distribution of chemicals. The power of LSER lies in its ability to deconstruct complex solvation phenomena into discrete, chemically meaningful interactions that can be quantified and predicted. This review serves as a technical guide to the computational resources available for implementing LSER methodologies, with particular emphasis on their application within broader research on solute descriptor determination and utilization.
The fundamental Abraham LSER model is expressed through two primary equations that quantify solute transfer between phases. For partitioning between two condensed phases, the model uses:
log(P) = cp + epE + spS + apA + bpB + vpVx [2] [1]
where P is the partition coefficient, and the lower-case coefficients (cp, ep, sp, ap, bp, vp) are system constants describing the complementary properties of the phases involved. For gas-to-solvent partitioning, the equation becomes:
log(KS) = ck + ekE + skS + akA + bkB + lkL [1]
In these equations, the capital letters represent the solute's molecular descriptors: Vx is McGowan's characteristic volume, L is the gas-liquid partition coefficient in n-hexadecane at 298 K, E represents excess molar refraction, S stands for dipolarity/polarizability, A characterizes hydrogen bond acidity, and B represents hydrogen bond basicity [2] [1]. The successful application of LSER methodology hinges on the accurate determination of these descriptors and system coefficients through appropriate computational tools and experimental protocols.
Foundational Concepts and Theoretical Framework
The Thermodynamic Basis of LSER
The remarkable linearity observed in LSER models, even for strong specific interactions like hydrogen bonding, finds its foundation in solvation thermodynamics. The process of solvation can be conceptually divided into an endoergic component (cavity formation and solvent reorganization) and exoergic components (solute-solvent attractive forces). The LSER framework successfully captures the net balance of these opposing energetic contributions through its linear free energy relationship [2] [1]. This thermodynamic basis ensures the model's applicability across diverse chemical systems and explains its predictive power for free-energy-related properties.
When applying LSERs to chromatographic retention, the retention factor (log k') is typically used as the free-energy-related property (SP in the general LSER equation). The coefficients in the LSER equation then reflect the difference in solvation properties between the mobile and stationary phases [2]. This interpretation allows researchers to extract meaningful chemical information about chromatographic systems, enabling rational method development in analytical chemistry and pharmaceutical analysis. The model's versatility extends to various chromatographic modes, including reversed-phase, normal-phase, and micellar electrokinetic capillary chromatography [2].
Chemical Interpretation of Solute Descriptors
Vx (McGowan's Characteristic Volume): This descriptor characterizes the solute's molecular size and represents the endoergic cost of cavity formation in the solvent. It is calculated from molecular structure and reflects the energy required to displace solvent molecules to accommodate the solute [2] [1].
L (Gas-Hexadecane Partition Coefficient): This experimental descriptor primarily reflects dispersive interactions between the solute and an alkane solvent, serving as a reference for van der Waals forces [1].
E (Excess Molar Refraction): Derived from refractive index data, this descriptor quantifies the solute's polarizability, particularly from n- or π-electrons. It helps capture interactions that arise from electron-rich regions in molecules [2] [1].
S (Dipolarity/Polarizability): This parameter represents the solute's ability to engage in dipole-dipole and dipole-induced dipole interactions. It encompasses both permanent and temporary polarization effects [2] [1].
A (Hydrogen Bond Acidity): A measure of the solute's ability to donate hydrogen bonds, this descriptor quantifies the strength of solute-to-solvent hydrogen bonding where the solute acts as the proton donor [2] [1].
B (Hydrogen Bond Basicity): This descriptor characterizes the solute's ability to accept hydrogen bonds, representing interactions where the solute acts as the proton acceptor [2] [1].
Computational Approaches for LSER Parameter Determination
Experimental Methodologies for Solute Descriptor Determination
The accurate determination of solute descriptors forms the foundation of reliable LSER applications. The following protocols outline standardized experimental approaches for measuring each descriptor:
Protocol for Vx Determination: McGowan's characteristic volume is calculated from molecular structure using the established formula based on atomic contributions and bond types. The calculation involves summing atomic volume parameters and subtracting a correction factor for molecular connectivity. This descriptor can be computed directly from molecular structure without experimental measurement, making it accessible for virtual screening applications [2] [1].
Protocol for L Determination: The L descriptor is experimentally determined as the logarithm of the gas-to-n-hexadecane partition coefficient at 298 K. Measurement is typically performed using gas-liquid chromatography with n-hexadecane as the stationary phase. The solute's retention time relative to an unretained compound provides the partition coefficient, with multiple determinations across different column loadings to ensure accuracy and independence of column characteristics [1].
Protocol for E Determination: The excess molar refraction is calculated from the solute's refractive index measured at 20°C for the sodium D line. The descriptor is computed using the formula: E = (n²D - 1)/(n²D + 2) - 0.1, where the 0.1 term represents the contribution of dispersive forces estimated from the alkane reference. For solids, the measurement requires dissolution in a suitable solvent and extrapolation to infinite dilution [1].
Protocol for S, A, and B Determination: These descriptors are typically determined simultaneously through a series of partition coefficient measurements in well-characterized systems. The recommended protocol involves:
- Measuring log P values for the solute in multiple solvent systems (e.g., water-organic solvent partitions)
- Utilizing gas-solvent partition coefficients (log K) for additional data points
- Performing multiparameter linear regression using established system coefficients
- Iteratively refining descriptors until consistent values are obtained across all solvent systems A minimum of 5-6 independent partition measurements is recommended for reliable determination of S, A, and B descriptors [2].
The following table summarizes the core LSER solute descriptors and their experimental determination methods:
Table 1: LSER Solute Descriptors and Experimental Determination Methods
| Descriptor | Molecular Property | Experimental Determination Method | Typical Range |
|---|---|---|---|
| Vx | Molecular size/volume | Calculation from molecular structure | 0.2 - 4.0 |
| L | Dispersive interactions | Gas-liquid chromatography in n-hexadecane | -0.5 - 8.0 |
| E | Polarizability | Refractive index measurement | 0.0 - 3.0 |
| S | Dipolarity/polarizability | Solvent partition coefficients | 0.0 - 2.5 |
| A | Hydrogen bond acidity | Solvent partition coefficients | 0.0 - 1.5 |
| B | Hydrogen bond basicity | Solvent partition coefficients | 0.0 - 2.0 |
Computational Tools for LSER Implementation
While specialized commercial software dedicated exclusively to LSER calculations is not prominently featured in current literature, researchers typically employ a combination of statistical, computational, and custom tools to implement LSER methodologies:
Statistical Software for Regression Analysis: The core computational requirement for LSER applications is multiple linear regression analysis to determine system coefficients or solute descriptors. Standard statistical packages including R, Python (with scikit-learn, statsmodels, or pandas libraries), MATLAB, and SAS are widely employed for this purpose. These tools facilitate the multiparameter linear least squares regression analysis necessary to correlate experimental partition coefficients with solute descriptors [2]. The regression models typically follow the form SP = c + eE + sS + aA + bB + vV for condensed phase partitions or SP = c + eE + sS + aA + bB + lL for gas-to-solvent partitions, where SP represents the free-energy-related property being studied [2] [1].
Quantum Chemical Computation: For researchers seeking to predict solute descriptors from molecular structure, quantum chemical calculations provide a valuable approach. Software packages such as Gaussian, Schrödinger Suite, and Spartan enable the computation of electronic properties that correlate with LSER descriptors. Molecular polarizability, dipole moments, and electrostatic potential surfaces can be derived from these calculations and used to estimate S, A, and B descriptors, though experimental validation remains essential [1].
Partial Solvation Parameters (PSP) Framework: An emerging approach for extracting thermodynamic information from LSER databases involves the Partial Solvation Parameters (PSP) framework. PSPs are designed with an equation-of-state thermodynamic basis that facilitates information exchange between LSER databases and molecular thermodynamics. This framework defines hydrogen-bonding PSPs (σa and σb) reflecting acidity and basicity characteristics, a dispersion PSP (σd) for weak dispersive interactions, and a polar PSP (σp) for Keesom-type and Debye-type polar interactions [1]. The PSP approach enables estimation of free energy change (ΔGhb), enthalpy change (ΔHhb), and entropy change (ΔShb) upon hydrogen bond formation, extending the utility of LSER-derived parameters.
Table 2: Software Tools for LSER Implementation and Their Applications
| Software Category | Representative Tools | LSER Application | Key Advantages |
|---|---|---|---|
| Statistical Analysis | R, Python (scikit-learn), MATLAB, SAS | Multiple linear regression for system coefficients | Flexibility, extensive statistical diagnostics, customization |
| Quantum Chemistry | Gaussian, Schrödinger Suite, Spartan | Prediction of solute descriptors from molecular structure | Ability to handle novel compounds without experimental data |
| Custom Spreadsheet Solutions | Microsoft Excel with LINEST function | Basic LSER regression for limited datasets | Accessibility, ease of use, visual data inspection |
| Database Management | Custom SQL databases, LSER published databases | Compilation of solute descriptors and system coefficients | Access to curated parameters for diverse compounds |
Advanced Applications and Emerging Methodologies
Integration with Machine Learning Approaches
Recent advances demonstrate the powerful synergy between LSER methodologies and machine learning (ML) for predictive modeling. While not directly applied to traditional LSER, the successful implementation of ML for predicting selective laser sintering 3D printing of drug products illustrates the potential for such approaches in physicochemical property prediction [64]. In this context, researchers achieved high prediction accuracy (F1 score of 88.9%) by combining multiple data modalities (FT-IR, XRPD, DSC) in a consensus model [64]. This multi-modal data integration approach suggests a promising pathway for enhancing LSER predictions, particularly for complex drug-like molecules where traditional descriptor determination proves challenging.
The workflow for machine learning-enhanced LSER modeling typically involves:
- Compiling experimental partition data for diverse solutes
- Calculating or measuring traditional LSER descriptors
- Generating additional molecular descriptors from computational chemistry
- Training ML models (e.g., random forest, gradient boosting, neural networks) to predict partition coefficients
- Validating models against external test sets This approach can capture non-linear relationships that may exist in complex biological partitioning systems, potentially extending beyond the traditional linear free-energy relationship framework while maintaining chemical interpretability through descriptor analysis.
LSER in Pharmaceutical Research and Drug Development
LSER methodologies find particularly valuable applications in pharmaceutical research, where predicting solute behavior in complex biological environments is essential for drug development:
Permeability Prediction: LSER models have been successfully applied to predict drug transport across biological membranes, including gastrointestinal absorption, blood-brain barrier penetration, and skin permeation. The descriptors provide mechanistic insight into the molecular characteristics governing permeability, guiding medicinal chemistry optimization.
Solubility and Formulation: The ability to predict solubility in various solvents and formulation matrices makes LSER invaluable for preformulation studies. The hydrogen bonding descriptors (A and B) are particularly informative for understanding drug-excipient interactions and potential compatibility issues.
Chromatographic Method Development: In analytical chemistry supporting pharmaceutical development, LSER guides the rational selection of chromatographic conditions by characterizing the interaction capabilities of stationary and mobile phases. This application significantly reduces method development time through systematic optimization [2].
The following diagram illustrates the integrated workflow for applying LSER methodologies in pharmaceutical research:
LSER Application Workflow in Pharmaceutical Research
Essential Research Reagent Solutions for LSER Studies
The experimental determination of LSER parameters requires specific chemical systems and reference materials. The following table details key research reagents and their functions in LSER studies:
Table 3: Essential Research Reagents for LSER Parameter Determination
| Reagent/System | Function in LSER Studies | Application Context |
|---|---|---|
| n-Hexadecane | Reference solvent for determining L descriptor using gas-liquid chromatography | Represents pure dispersive interactions without polar or hydrogen bonding contributions |
| Water-Solvent Partition Systems | Determination of S, A, B descriptors through measured partition coefficients | Multiple solvent systems (e.g., octanol-water, alkane-water, chloroform-water) provide diverse interaction environments |
| Reference Solutes | Calibration of system coefficients in LSER equations | Compounds with well-established descriptor values for method validation |
| Chromatographic Phases | Characterization of stationary phase properties through retention data | Reversed-phase, normal-phase, and specialized HPLC columns for chromatographic LSER |
| Gas Chromatography Systems | Measurement of gas-solvent partition coefficients (log K) | Determination of L descriptor and additional data points for S, A, B refinement |
LSER methodology continues to provide invaluable insights into molecular interactions across chemical, pharmaceutical, and environmental sciences. While the computational tools for implementing LSER are primarily adapted from general statistical and quantum chemical software, the robust theoretical foundation ensures continued relevance and application. The ongoing development of approaches like Partial Solvation Parameters (PSP) promises enhanced extraction of thermodynamic information from existing LSER databases [1]. For researchers engaged in solute descriptor determination and application, mastery of both experimental protocols and computational implementation remains essential for generating reliable, interpretable results.
Future developments in LSER computational tools will likely focus on enhanced integration with machine learning approaches, improved prediction of descriptors solely from molecular structure, and expansion to more complex biological partitioning systems. The integration of multi-modal data, as demonstrated in pharmaceutical 3D printing applications [64], suggests a promising pathway for increasing prediction accuracy while maintaining the chemical interpretability that has made LSER methodology enduringly valuable across scientific disciplines. As these computational approaches evolve, LSER will continue to provide fundamental insights into solute-solvent interactions, supporting rational design in drug development and molecular sciences.
Overcoming LSER Challenges: Troubleshooting Descriptor Determination and Model Optimization
Common Pitfalls in Experimental Descriptor Determination and Strategies for Mitigation
The accurate determination of experimental descriptors, such as those used in Linear Solvation Energy Relationship (LSER) studies denoted as Vx, E, S, A, B, L, is a cornerstone of modern physicochemical property prediction and drug development. These descriptors quantitatively represent molecular properties including excess molar refraction (E), dipolarity/polarizability (S), hydrogen-bond acidity (A), hydrogen-bond basicity (B), and the logarithm of the gas-hexadecane partition coefficient (L). The reliability of models predicting solubility, permeability, and toxicity in pharmaceutical research depends fundamentally on the accuracy of these experimentally derived descriptors. However, the process of determining these values is fraught with methodological challenges that can compromise data integrity and model performance if not properly addressed.
Researchers must navigate a complex landscape of experimental and computational pitfalls throughout the descriptor determination process. From initial experimental design to final data validation, each stage introduces potential sources of error that can systematically bias results. This technical guide examines the most prevalent pitfalls in experimental descriptor determination, provides evidence-based mitigation strategies, details essential experimental protocols, and offers practical tools for implementation within research and development settings. By addressing these challenges systematically, scientists can enhance the reliability of their descriptor data and improve the predictive power of subsequent LSER models in drug development applications.
Common Pitfalls and Mitigation Strategies
The determination of experimental descriptors involves multiple critical stages where systematic errors can be introduced. Understanding these pitfalls and implementing appropriate countermeasures is essential for generating reliable, reproducible descriptor data.
Table 1: Common Pitfalls in Experimental Descriptor Determination and Corresponding Mitigation Strategies
| Pitfall Category | Specific Pitfall | Impact on Descriptor Accuracy | Recommended Mitigation Strategy |
|---|---|---|---|
| Experimental Design | Insufficient sample size | Reduced statistical power; unreliable descriptor values [65] | Perform a priori power analysis; use statistical tools for sample size calculation [65] |
| Neglecting confounding variables | Systematic bias in descriptor measurements [65] | Conduct thorough literature review; implement control groups; use statistical control methods [65] | |
| Selection bias in compound choice | Non-representative descriptor values; limited applicability domains | Apply random sampling techniques; use stratified selection based on chemical space [65] | |
| Measurement Approach | Improper control groups | Inaccurate baseline measurements affecting calculated descriptors [65] | Implement appropriate control types (placebo, no-treatment, wait-list) based on experiment [65] |
| Instrumental error | Systematic measurement inaccuracies [66] | Regular equipment calibration; controlled environmental conditions; proper staff training [66] | |
| Solvent-accessible surface miscalculation | Errors in S descriptor determination [67] | Use validated algorithms (Neighbor Vector); account for spatial orientation of atoms [67] | |
| Data Analysis | Multiple testing without correction | Increased false discovery rate for significant descriptors [68] | Apply Bonferroni or Benjamini-Hochberg procedures; limit metrics to essential ones [68] |
| Peeking and early stopping | Inflated false positives; biased descriptor values [68] | Pre-determine sample size; avoid interim analysis; use sequential testing if needed [68] | |
| Mishandling of outliers | Distorted descriptor averages and relationships [69] | Investigate outlier causes; use Winsorization or robust statistics [69] |
Pitfall Analysis in Experimental Design
Insufficient sample size represents one of the most prevalent pitfalls in experimental descriptor determination. Underpowered studies increase the risk of Type II errors (false negatives), produce unreliable effect size estimates, and limit the generalizability of findings [65]. This is particularly problematic for descriptor determination where establishing precise values requires sufficient statistical power to detect meaningful effects. For LSER applications, insufficient sampling across chemical space can lead to descriptors that fail to accurately predict properties for compound classes not included in the training set.
The neglect of confounding variables presents another significant challenge in descriptor determination. Confounders are variables that correlate with both the independent and dependent variables, potentially leading to spurious associations [65]. In the context of LSER descriptor measurement, factors such as temperature fluctuations, solvent impurities, or measurement timing can act as confounders that systematically bias results. For example, inaccurate control of temperature during solubility measurements can directly impact the determination of partition coefficients central to descriptor calculation.
Selection bias in compound selection can severely limit the applicability domain of subsequently developed LSER models. When compounds selected for descriptor measurement do not adequately represent the chemical space of interest, the resulting models will have limited predictive utility for novel compounds. This bias often arises from convenience sampling of readily available compounds rather than strategic selection based on diverse molecular features.
Measurement and Data Analysis Challenges
Instrumental errors introduce systematic inaccuracies in descriptor measurements and can arise from using outdated, faulty, or improperly calibrated equipment [66]. For descriptor determination relying on spectroscopic measurements or chromatographic retention times, even minor instrumental drift can significantly impact results. These errors are particularly pernicious as they may not be readily apparent in the data but can systematically bias descriptor values.
The accurate computation of solvent-accessible surface area (SASA) presents specific challenges for the determination of S descriptors representing dipolarity/polarizability. SASA is a geometric measure of atomic exposure to solvent that influences solvation energy [70]. Traditional SASA calculation methods, such as the Shrake-Rupley algorithm which involves rolling a spherical probe around a molecular surface, are computationally demanding and not pair-wise decomposable [67]. This makes them impractical for high-throughput descriptor calculation where thousands of compounds must be evaluated.
Multiple testing problems emerge when researchers evaluate numerous potential relationships without statistical correction, increasing the probability of false discoveries [68]. In descriptor determination, this might involve testing multiple functional forms or parameter combinations until statistically significant relationships are found. Without proper correction, the resulting descriptors may capture noise rather than meaningful physicochemical relationships, compromising model validity.
Experimental Protocols for Descriptor Validation
Protocol for Solvent-Accessible Surface Area Determination
The accurate determination of solvent-accessible surface area is critical for calculating S descriptors in LSER systems. This protocol outlines a standardized approach for SASA calculation suitable for descriptor determination.
Principle: The SASA represents the surface area of a biomolecule that is accessible to a solvent molecule, typically modeled using a spherical probe with radius of 1.4Å (approximating a water molecule) [70]. The extent to which an amino acid interacts with its environment is proportional to its exposure to these environments, making SASA a geometric measure of this exposure [67].
Materials and Equipment:
- High-performance computing workstation
- Molecular structure files (PDB format recommended)
- Specialized software (MSMS algorithm or Neighbor Vector implementation)
Procedure:
- Structure Preparation: Obtain or generate optimized 3D molecular structures for all compounds under investigation. Ensure structures include hydrogen atoms and represent relevant protonation states at physiological pH.
- Parameter Selection: Set probe radius to 1.4Å to represent water molecules. Define van der Waals radii according to established force field parameters (e.g., AMBER, CHARMM).
- Algorithm Selection: For high-throughput applications, implement the "Neighbor Vector" algorithm which provides optimal balance between computational speed and accuracy [67]. For maximum accuracy in smaller datasets, use the MSMS algorithm with default parameters.
- Calculation Execution: Process molecular structures through selected algorithm. For the Neighbor Vector method, ensure proper accounting of spatial orientation of neighboring atoms to address shortcomings of simple burial approximations.
- Validation: Compare results with known standards or repeat calculations using alternative methods to verify accuracy.
Data Analysis: Express SASA values in units of Ų per molecule or Ų per residue for larger compounds. Normalize values by total molecular surface area for comparative analyses. Incorporate calculated SASA values into S descriptor determination using established LSER equations.
Protocol for Hydrophobicity Descriptor Determination
Principle: Hydrophobic interactions are a fundamental driving force in many chemical and biological phenomena and are represented by multiple descriptors in LSER systems [71]. This protocol standardizes the determination of hydrophobicity-related descriptors through experimental transfer free energy measurements.
Materials and Equipment:
- High-purity water (HPLC grade)
- Organic solvents (n-octanol, hexadecane, etc.)
- Analytical balance (precision ±0.0001 g)
- Temperature-controlled shaking incubator
- HPLC system with UV detection
- Glass vials with PTFE-lined caps
Procedure:
- Solution Preparation: Prepare saturated solutions of test compounds in aqueous and organic phases by adding excess compound to each solvent system.
- Equilibration: Agitate mixtures continuously for 24 hours in a temperature-controlled environment (25.0°C ± 0.1°C) to ensure proper equilibration.
- Phase Separation: Allow phases to separate completely (minimum 6 hours at constant temperature).
- Concentration Analysis: Quantify compound concentrations in both phases using validated HPLC-UV methods with appropriate calibration standards.
- Partition Coefficient Calculation: Calculate partition coefficients (P) as the ratio of compound concentration in the organic phase to that in the aqueous phase.
- Descriptor Derivation: Convert partition coefficients to L descriptors using established LSER equations.
Data Analysis: Determine L descriptors as logP values for the gas-hexadecane system. Perform replicate measurements (minimum n=5) to establish precision. Include reference compounds with known descriptor values to validate methodological accuracy.
Figure 1: Experimental descriptor determination workflow showing key stages with associated pitfalls and mitigation strategies integrated throughout the process.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Essential Research Reagents and Materials for Experimental Descriptor Determination
| Reagent/Material | Specification | Function in Descriptor Determination | Quality Control Requirements |
|---|---|---|---|
| Reference Compounds | USP/EP grade with certificate of analysis | Method validation and descriptor calibration | Purity ≥98%; structural confirmation via NMR/MS |
| Chromatographic Solvents | HPLC grade, low UV cutoff | Mobile phase for partition coefficient determination | Lot-to-lot consistency testing; filtration through 0.22μm membrane |
| Buffer Components | ACS grade, ≥99% purity | pH control in hydrogen-bonding descriptor studies | pH verification ±0.01 units; conductivity testing |
| Solid Phase Extraction Cartridges | C18 or appropriate chemistry | Sample cleanup before analytical determination | Recovery validation for compound classes of interest |
| Calibration Standards | Traceable to reference standards | Instrument calibration and response verification | Documentation of uncertainty and traceability |
| Molecular Modeling Software | Validated algorithms (e.g., Neighbor Vector) | Computational descriptor calculation (SASA, etc.) [67] | Benchmark against experimental data |
Advanced Methodologies for Specific Descriptor Classes
Hydrogen-Bonding Descriptor (A and B) Determination
The accurate quantification of hydrogen-bond acidity (A) and basicity (B) presents particular challenges in LSER descriptor determination due to the complex nature of hydrogen-bonding interactions and their sensitivity to experimental conditions.
Spectroscopic Protocol for A and B Descriptors:
- Principle: Hydrogen-bonding strength can be quantified through spectroscopic shifts observed when compounds interact with carefully selected reference partners in controlled solvent environments.
- Materials: Deuterated solvents (CDCl₃, DMSO-d₆), reference hydrogen-bond acceptors/donors (e.g., 4-fluorophenol for basicity measurement), NMR spectrometer.
- Procedure:
- Prepare solutions with fixed concentration of reference compound and varying concentrations of test compound.
- Record chemical shift changes for reference proton (e.g., hydroxyl proton of 4-fluorophenol).
- Construct association curves plotting chemical shift versus molar ratio.
- Calculate association constants from nonlinear regression of binding curves.
- Data Analysis: Convert association constants to A and B descriptors using established transfer functions from the literature. Account for solvent effects using appropriate correction factors.
The hydrogen-bonding descriptors are particularly susceptible to solvent effects and concentration dependencies. Research indicates that structural competition between interfacial and bulk water significantly influences hydrophobic interactions [71], which must be considered when determining A and B descriptors in aqueous environments. Recent advances in our understanding of hydrogen bonding networks suggest that directional nature and temperature effects must be standardized across determinations to ensure descriptor comparability.
Dipolarity/Polarizability (S) Descriptor Refinement
The S descriptor representing dipolarity/polarizability can be refined through multiple complementary approaches to enhance accuracy:
Computational Refinement Protocol:
- Electronic Structure Calculations: Perform density functional theory (DFT) calculations to obtain dipole moment and polarizability tensors.
- Solvent Effect Modeling: Use polarized continuum models to account for solvent effects on molecular polarizability.
- Experimental Correlation: Validate computational results against experimentally determined chromatographic retention parameters in multiple solvent systems.
- Descriptor Integration: Combine computational and experimental values using Bayesian integration to obtain final S descriptor with uncertainty estimates.
This multimodal approach addresses the limitation of single-method determinations and provides robust S descriptors with quantified uncertainty. The computational aspects benefit from recent advances in SASA approximation methods, particularly the "Neighbor Vector" algorithm which provides an optimal balance between computational speed and accuracy for assessing solvent exposure effects on polarizability [67].
Figure 2: Multimodal approach to descriptor determination combining experimental and computational methods for enhanced accuracy and reliability.
The accurate determination of LSER solute descriptors (Vx, E, S, A, B, L) requires meticulous attention to experimental design, measurement protocols, and data analysis practices. The pitfalls discussed throughout this guide—from insufficient sample sizes to improper handling of confounding variables—represent significant threats to descriptor accuracy and the predictive performance of subsequent models. By implementing the systematic mitigation strategies outlined, researchers can significantly enhance the reliability of their experimental descriptor determinations.
A proactive systems approach that anticipates potential errors rather than simply reacting to them creates a foundation for robust descriptor development [66]. This includes establishing standardized protocols, implementing regular equipment calibration, fostering open communication about methodological challenges, and applying appropriate statistical corrections throughout the analysis process. Furthermore, the integration of computational and experimental approaches provides a powerful strategy for descriptor validation and refinement.
As pharmaceutical research continues to demand more accurate property prediction models for drug development, the rigorous determination of experimental descriptors remains fundamentally important. By addressing the common pitfalls through the methodologies detailed in this guide, researchers can generate descriptor values with greater confidence, ultimately supporting the development of more reliable predictive models in pharmaceutical sciences and beyond.
Addressing Molecular Conformation Complexity in Vx and S Parameter Calculation
Within the framework of Linear Solvation Energy Relationship (LSER) research, the solute descriptors Vx (McGowan's characteristic molar volume) and S (dipolarity/polarizability) are foundational for predicting a molecule's partitioning behavior in chemical and biological systems [72]. Traditional LSER models often treat these descriptors as fixed values, typically derived from a single, low-energy molecular conformation. However, molecules in solution are dynamic entities that sample an ensemble of three-dimensional conformations accessible at finite temperature [73]. This conformational flexibility directly influences key molecular properties; for instance, the radius of gyration (Rg) and persistence length (lp) of a polymer chain—properties sensitive to conformation—can be measured experimentally by techniques like Small Angle X-Ray Scattering (SAXS) [74]. The core challenge addressed in this guide is that a molecule's effective Vx and S, and thus its observed LSER behavior, are not determined by a single structure but by a Boltzmann-weighted average over its entire conformational ensemble. Failure to account for this complexity can introduce significant error into property predictions, especially for flexible pharmaceuticals and disordered biomolecules [75] [76]. This guide provides detailed methodologies for calculating Vx and S parameters that accurately reflect this conformational reality.
Computational Approaches for Conformational Sampling
A robust computational workflow is essential for generating a representative set of molecular conformations.
Conformer Generation with CREST
The CREST program (Conformer-Rotamer Ensemble Sampling Tool) represents the current state-of-the-art for exhaustive conformational sampling [73]. It utilizes the semi-empirical extended tight-binding method (GFN2-xTB) to calculate energies, offering a favorable balance between accuracy and computational cost.
Detailed Protocol:
- Input Preparation: Provide the molecular structure of the compound of interest as a 3D coordinate file (e.g., .xyz or .pdb).
- CREST Execution: Run CREST with the default settings to initiate a metadynamics-based conformational search in vacuum. For biologically relevant predictions, a follow-up calculation using an implicit solvent model (e.g., ALPB for water) is critical.
- Output Analysis: CREST returns a conformational ensemble comprising multiple unique structures, each annotated with its relative energy (Ei) and a statistical weight (p_i^CREST) derived from a quasi-harmonic free energy calculation [73]:
p_i^CREST = [ d_i * exp(-E_i / k_B * T) ] / [ Σ_j d_j * exp(-E_j / k_B * T) ]whered_iis the degeneracy of the conformer,k_Bis Boltzmann's constant, andTis the temperature.
Advanced Sampling and Refinement
For higher accuracy, the CREST-generated ensembles can be refined.
- DFT Single-Point Calculations: Perform higher-level energy calculations (e.g., using Density Functional Theory, DFT) on the CREST-derived conformers to obtain more accurate electronic energies [73].
- Boltzmann Weighting: Recalculate the statistical weights for each conformer using the more accurate DFT energies, which supersede the semi-empirical weights for final property averaging.
Table 1: Software Tools for Conformational Analysis
| Tool | Primary Function | Key Advantage | Reference |
|---|---|---|---|
| CREST | Conformer-Rotamer Ensemble Generation | Uses metadynamics & semi-empirical DFT for exhaustive sampling | [73] |
| GFN2-xTB | Semi-empirical Energy Calculation | Fast and accurate, accounts for electronic effects | [73] |
| DFT (e.g., Gaussian, ORCA) | High-Quality Energy Calculation | Provides benchmark-quality energies for refinement | [73] |
| RDKit | Cheminformatics Toolkit | Rule-based or stochastic conformer generation (less exhaustive) | [73] |
Calculating Ensemble-Averaged Vx and S Descriptors
Once a representative, weighted conformational ensemble is generated, the molecular descriptors must be calculated for each conformer and averaged.
McGowan's Characteristic Molar Volume (Vx)
The Vx descriptor is proportional to the molecular volume, which is highly sensitive to conformational changes [72].
Methodology:
- Per-Conformer Volume Calculation: For each conformer in the ensemble, calculate its molecular volume. This can be achieved by placing the molecule in a virtual box of solvent probes and counting the number of occupied grid points, or by using algorithms that compute the volume enclosed by the van der Waals surface.
- Ensemble Averaging: Calculate the Boltzmann-weighted average volume across the entire ensemble. The final, conformationally-aware Vx descriptor is then derived from this averaged volume.
Dipolarity/Polarizability (S) Descriptor
The S parameter reflects a molecule's ability to engage in dipole-dipole and induced dipole interactions, both of which are functions of the 3D electronic structure [72].
Methodology:
- Electronic Property Calculation: For each conformer, compute its dipole moment and polarizability tensor using quantum chemical methods (e.g., DFT). The magnitude of the dipole moment is a direct contributor to the S descriptor.
- Averaging of Properties: The effective S descriptor for the ensemble is not a simple average of individual S values. Instead, the dipole moment vectors and polarizability tensors must be averaged according to their Boltzmann weights to account for the net polarizability and the magnitude and direction of the average dipole moment across all populated states.
Experimental Validation of Conformationally-Aware Descriptors
Computational predictions require experimental validation. Several techniques can probe conformational properties that influence Vx and S.
Small-Angle X-Ray Scattering (SAXS)
SAXS is a powerful, label-free technique for studying the overall shape and structural transitions of macromolecules in solution [77] [74] [76]. It is particularly useful for flexible systems like intrinsically disordered proteins and single-stranded nucleic acids [76].
Experimental Protocol:
- Sample Preparation: Prepare a monodisperse solution of the target molecule in an appropriate buffer. A matched buffer blank must also be prepared for background subtraction.
- Data Collection: Expose the sample and blank to a monochromatic X-ray beam and collect scattered intensity at low angles (typically 0-5 degrees). The scattering vector is defined as
q = 4π sin(θ) / λ, where2θis the scattering angle andλis the X-ray wavelength [77]. - Data Analysis:
- Subtract the buffer scattering from the sample scattering to obtain the net scattering profile of the solute, I(q).
- Analyze the Guinier region (at low q) to determine the radius of gyration (Rg), which describes the overall size of the molecule [77] [74].
- Compute the pair-distance distribution function, p(r), via an indirect Fourier transform of I(q). The p(r) function provides a real-space representation of the particle's shape and conformation [77].
Escape-Time Electrometry (ETe)
ETe is an emerging technique that measures the effective electrical charge (q_eff) of a molecule in solution with high precision [74]. Since charge renormalization is a function of the 3D conformation of the molecular charge distribution, ETe provides a sensitive, orthogonal measure of molecular shape.
Experimental Protocol:
- Confinement: Charged molecules are spatially confined to diffuse within a slit formed by two like-charged parallel surfaces.
- Measurement: The time individual molecules spend in this electrostatic trap (escape time) is measured, typically via optical microscopy.
- Analysis: The escape time distribution is used to infer the molecule-surface interaction energy, which is parametrized to yield the effective charge, qeff [74]. This measured qeff can be directly compared with values calculated from conformational ensembles (e.g., using Poisson-Boltzmann theory) to validate the models [74].
Table 2: Experimental Techniques for Conformational Validation
| Technique | Measured Parameter | Relation to Vx/S | Application Note |
|---|---|---|---|
| SAXS | Rg, p(r), molecular shape | Rg correlates with molecular volume (Vx); shape influences polarizability (S) | Ideal for flexible proteins, nucleic acids, and polymers [74] [76] |
| Escape-Time Electrometry (ETe) | Effective charge (q_eff) | Sensitive to 3D charge distribution, which is linked to conformation and S | Single-molecule precision; useful for polyelectrolytes like nucleic acids [74] |
| Molecular Dynamics (MD) | Theoretical Rg, q_eff, energy | Provides atomic-level trajectories for direct comparison with SAXS/ETe | Used synergistically with SAXS (MD-SAXS) to model kinetics [77] |
Workflow for Conformation-Aware Descriptor Determination
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Computational Resources
| Item / Resource | Function / Application |
|---|---|
| CREST Software | Open-source tool for generating conformer-rotamer ensembles via metadynamics [73]. |
| GFN2-xTB Hamiltonian | Semi-empirical quantum mechanical method for accurate and rapid energy calculations during sampling [73]. |
| GEOM Dataset | Large-scale dataset containing high-quality conformers for over 450,000 molecules; useful for benchmarking [73]. |
| Monodisperse Protein/NA Sample | High-purity, aggregated-free sample essential for reliable SAXS data collection [76]. |
| Synchrotron SAXS Beamline | High-intensity X-ray source enabling time-resolved SAXS studies on flexible systems [77] [76]. |
| Implicit Solvent Model (ALPB/GB) | Computational model to simulate solvation effects during conformational sampling and energy evaluation [73]. |
| LSER Solute Descriptor Database | Curated database (e.g., UFZ-LSER) for obtaining descriptor values for validation [72]. |
Accurately calculating Vx and S parameters in the face of molecular conformational complexity is a multi-faceted problem requiring an integrated computational and experimental strategy. The methodologies outlined in this guide—from exhaustive conformational sampling with CREST and DFT refinement to experimental validation with SAXS and ETe—provide a robust framework for moving beyond single-structure approximations. By explicitly accounting for the conformational ensemble, researchers can develop more predictive and physiologically relevant LSER models, ultimately enhancing efforts in rational drug design and materials science.
Linear Solvation Energy Relationships (LSERs) are a fundamental tool for understanding the intermolecular interactions governing chemical processes, including chromatographic retention. The widely accepted Abraham LSER model is represented by the equation:
SP = c + eE + sS + aA + bB + vV [2]
In this model, the solute descriptors represent specific molecular properties: V represents molecular size, E represents polarizability, S represents dipolarity, and A and B represent hydrogen-bond acidity and basicity, respectively [2]. For strongly ionizable compounds, the assignment of the hydrogen-bonding descriptors A and B becomes particularly challenging because the ionization state of a molecule can dramatically alter its hydrogen-bonding character. A compound that is neutral at one pH may become ionized at physiological or chromatographic pH levels, fundamentally changing its ability to donate or accept hydrogen bonds [78]. This technical guide examines the specific challenges in assigning A and B descriptors for ionizable compounds and provides frameworks for researchers working within LSER-based investigations.
Theoretical Foundation: The LSER Framework and Ionizable Compounds
Fundamental LSER Principles
The LSER model mathematically represents the contribution of different intermolecular interactions to a free-energy related property (SP), which in chromatography is typically the log of the retention factor (log k') [2]. The coefficients (e, s, a, b, v) in the LSER equation are system-dependent and reflect the relative importance of each interaction in the chemical process being studied. The solute descriptors (E, S, A, B, V) are postulated to be temperature-independent and fundamentally reflect the solute's intrinsic ability to engage in the various interactions [2].
For partition processes between two condensed phases, such as in reversed-phase liquid chromatography, the LSER mathematically represents the difference between the solute's interactions with the two phases [2]. This theoretical foundation becomes significantly more complex when dealing with ionizable compounds whose effective interaction abilities change with pH.
The Ionization Challenge
Strongly ionizable compounds can undergo protonation or deprotonation in response to the pH of their environment. This change in ionization state profoundly affects their hydrogen-bonding capabilities:
- Acidic compounds upon deprotonation gain hydrogen-bond accepting capacity (increased B) while losing hydrogen-bond donating capacity (decreased A)
- Basic compounds upon protonation gain hydrogen-bond donating capacity (increased A) while losing hydrogen-bond accepting capacity (decreased B) [78]
The microspecies distribution (the relative abundance of different ionization states of the same parent molecule) at a given pH follows the Henderson-Hasselbalch equation [78]. For compounds with multiple ionizable groups, the situation becomes increasingly complex, with multiple microspecies potentially coexisting at a given pH, each with its own hydrogen-bonding characteristics.
Table 1: Impact of Ionization on Solute Descriptors
| Ionization Type | Effect on A Descriptor | Effect on B Descriptor | Overall Impact |
|---|---|---|---|
| Acid Dissociation | Decreases significantly | Increases significantly | Net hydrogen-bonding character changes substantially |
| Base Association | Increases significantly | Decreases significantly | Complete reversal of hydrogen-bonding profile possible |
| Multiple Ionizations | Complex, non-linear changes | Complex, non-linear changes | Descriptor assignment becomes pH-dependent |
Experimental Methodologies for Descriptor Assessment
Chromatographic Approaches for Ionizable Compounds
Reversed-phase high-performance liquid chromatography (RP-HPLC) with controlled mobile phase composition and pH provides a powerful experimental system for studying ionizable compounds. The retention of ionizable solutes depends on both mobile phase composition and pH, following relationships that can be modeled using approaches such as artificial neural networks (ANNs) [79].
For ionizable pesticides such as phenoxy acid herbicides (pKa range 2.3-4.3), retention modeling must simultaneously account for mobile phase composition (% acetonitrile ranging 30-70%) and pH (ranging 2-5) [79]. The effective mobile phase acidity and solute ionization constant both vary with co-solvent content, adding another layer of complexity to descriptor assignment.
The following workflow illustrates the experimental determination of descriptors for ionizable compounds:
High-Throughput Prediction Methods
With advances in computational power, high-throughput prediction of ionization equilibria has become feasible for thousands of compounds. These methods use Ionizable Atom Type (IAT) classifications, which are specific configurations of atoms within a chemical that have the propensity to protonate or deprotonate [78].
Probability distributions of pKa values for each IAT can be generated based on predictions for large chemical libraries (e.g., 32,413 compounds including 8,132 pharmaceuticals) [78]. This approach enables sensitivity analysis of predicted properties like volume of distribution (Vdss) on predicted pKa using Monte Carlo methods, acknowledging the uncertainty in descriptor assignment for ionizable compounds.
Table 2: Methodologies for Studying Ionizable Compounds
| Methodology | Key Features | Applications in Descriptor Assignment | Limitations |
|---|---|---|---|
| RP-HPLC with pH control | Direct experimental measurement across pH conditions; uses water-acetonitrile mobile phases [79] | Enables observation of how retention changes with ionization state | Requires careful control of mobile phase pH and composition effects on pKa |
| QSRR with WHIM/GETAWAY descriptors | Uses 3D molecular descriptors; combined with mobile phase attributes in ANN models [79] | Models retention without explicit solvatochromic descriptors | Descriptors may not fully capture ionization effects |
| High-throughput pKa prediction | Uses Ionizable Atom Types (IATs); probabilistic approach; suitable for large chemical libraries [78] | Provides rapid estimation of ionization state across pH range | Uncertainty in predictions requires Monte Carlo sensitivity analysis |
| LSER global models | Extends LSER to include pH-dependent term for dissociation degree [79] | Directly incorporates ionization into retention modeling | Requires knowledge of solvatochromic descriptors |
Practical Applications in Pharmaceutical Research
Predicting Pharmacokinetic Behavior
The accurate assignment of A and B descriptors for ionizable compounds is crucial in pharmaceutical research for predicting tissue distribution, a key aspect of pharmacokinetics (PK). Chemical distribution within the body is heavily influenced by three key parameters: binding to tissue and plasma, hydrophobicity, and ionization [78].
For ionizable compounds, the tissue-plasma distribution coefficient (logD) depends on the ionization state, which varies with physiological pH. This relationship is particularly important for predicting the apparent volume of distribution at steady state (Vdss), a critical PK parameter [78]. The failure to properly account for ionization effects on hydrogen-bond descriptors can lead to significant errors in predicting tissue distribution.
Case Study: NHANES Chemicals Analysis
A study of 22 compounds monitored in human blood and serum by NHANES demonstrated the practical importance of accurate ionization modeling. Of these 22 compounds, 8 were predicted to be ionizable at physiological pH. For 5 of these 8 compounds, predictions based on ionization states were significantly different from predictions assuming neutral compounds [78]. This highlights how proper accounting of ionization effects on molecular descriptors leads to materially different predictions of pharmacokinetic behavior.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Tools for Ionizable Compound Research
| Tool/Category | Specific Examples | Function in Descriptor Assignment | Critical Considerations |
|---|---|---|---|
| pKa Prediction Software | ChemAxon, SPARC, ADMET Predictor [78] | Predicts ionization equilibria; identifies ionizable atom types | Different algorithms may give varying predictions; validation recommended |
| Molecular Descriptors | WHIM, GETAWAY descriptors [79] | Encodes 3D structural information relevant to retention | Less empirically derived than solvatochromic parameters |
| Chromatographic Systems | RP-HPLC with C18 columns; water-acetonitrile mobile phases [79] | Provides experimental retention data across pH conditions | Mobile phase composition affects apparent pKa; must be controlled |
| QSAR/QSPR Databases | DSSTox, CPCat, ACToR [78] | Provides reference data for model development and validation | Data quality varies; careful curation essential |
| Regression Tools | Artificial Neural Networks (ANNs), Genetic Algorithms [79] | Builds models relating structure to retention across multiple conditions | ANNs can handle non-linearity without presupposed relationship forms |
The assignment of A and B descriptors for strongly ionizable compounds remains a significant challenge in LSER research due to the pH-dependent nature of hydrogen-bonding characteristics. Successful approaches combine experimental chromatographic data across multiple pH conditions with computational predictions of ionization equilibria. The use of high-throughput pKa prediction methods, coupled with sensitivity analysis, provides a framework for addressing the uncertainty inherent in descriptor assignment for these compounds. As pharmaceutical research increasingly deals with ionizable molecules, the accurate characterization of their hydrogen-bonding descriptors becomes ever more critical for predicting pharmacokinetic behavior and environmental fate.
Optimizing Computational Methods for Accurate E and L Descriptor Prediction
Linear Solvation Energy Relationships (LSERs), also known as the Abraham model, provide a powerful quantitative framework for predicting a vast array of physicochemical properties and biochemical partitioning processes crucial to environmental science and drug development [80] [1]. The model's predictive power resides in its solute descriptors, which are numerical representations of a molecule's capacity for specific types of intermolecular interactions. The core model is expressed as log P = c + eE + sS + aA + bB + vV for partitioning between two condensed phases and log K = c + eE + sS + aA + bB + lL for gas-to-condensed phase partitioning [80] [1] [81]. While all descriptors are important, the excess molar refraction (E) and the gas-hexadecane partition coefficient (L) play distinct and critical roles.
The E descriptor represents a solute's excess molar refraction, which quantifies its ability to engage in van der Waals interactions, specifically through π- and n-electron pairs [80] [1]. The L descriptor is defined as the logarithm of the gas-hexadecane partition coefficient at 298 K and effectively characterizes a solute's hydrophobicity and its capacity for dispersion interactions [1]. Accurate prediction of these descriptors is therefore foundational for applying LSER models to the thousands of chemicals for which experimental data is unavailable, thus enabling reliable forecasts of solubility, permeability, and bioaccumulation potential in pharmaceutical and environmental research [80].
Computational Prediction Methods: Performance Comparison
Traditional methods for predicting E and L descriptors have relied on fragment-based Quantitative Structure-Property Relationship (QSPR) models, such as those implemented in the LSERD online database and the commercial software ACD/Absolv [80]. However, these methods can struggle with larger, more complex chemical structures featuring multiple functional groups [80]. The field has now been significantly advanced by the development of deep learning and other machine learning (ML) approaches, which offer superior handling of molecular complexity.
Table 1: Performance Comparison of Computational Methods for Solute Descriptor Prediction
| Prediction Method | Model Type | Key Features | Reported RMSE (E, L, or Overall) | Applicability Domain |
|---|---|---|---|---|
| QSPR (LSERD, ACD/Absolv) | Group Contribution | Fragmental approach | Overall RMSE ~1.0 log unit for properties like Kow [80] | Problematic for large, complex structures [80] |
| Deep Neural Networks (DNN) [80] | Deep Learning | Graph representations; Singletask & Multitask learning | RMSE range of 0.11-0.46 for different descriptors [80] | Better for complex structures; complementary to QSPR [80] |
| AbraLlama-Solute [81] | Fine-tuned Large Language Model (LLM) | Based on ChemLLaMA; inputs SMILES strings | High accuracy, comparable to existing methods [81] | Broad applicability for organic molecules |
As illustrated in Table 1, modern ML methods like DNNs and fine-tuned LLMs achieve high accuracy. The DNN models developed by Ulrich and Ebert demonstrated Root Mean Square Errors (RMSEs) ranging between 0.11 and 0.46 across the different solute descriptors, a significant level of precision [80]. Their research indicated that singletask models, trained on one descriptor at a time, outperformed multitask models on their dataset, likely due to the dataset's size [80]. Furthermore, they employed data augmentation strategies based on molecular tautomers to improve the training of their deep neural networks [80].
The AbraLlama Workflow: A Case Study in Modern Descriptor Prediction
The AbraLlama framework exemplifies the cutting edge of descriptor prediction by leveraging a fine-tuned Large Language Model (LLM), ChemLLaMA, which is specifically adapted for cheminformatics tasks [81]. The following diagram outlines the end-to-end workflow for developing and using such a model.
Model Development and Application Workflow
Detailed Experimental Protocol: Developing a Deep Learning Model for Descriptors
For researchers aiming to implement a DNN model for E and L prediction, the following protocol, based on the work of Ulrich and Ebert, provides a detailed roadmap [80].
1. Dataset Curation:
- Source: Begin with a known dataset, such as the Abraham Absolv dataset comprising 7,881 chemicals.
- Filtering: Refine the dataset by removing chemicals with missing core descriptors (e.g., the S descriptor), organometallics, and gases. This process resulted in a final curated set of 6,364 compounds for model development [80].
- Error Checking: Manually identify and correct obvious errors in molecular structures and descriptor values.
2. Model Architecture and Training:
- Representation: Convert molecular structures into a graph representation that encodes atoms and bonds, making it suitable for graph neural networks.
- Model Design: Develop both singletask (predicting one descriptor) and multitask (predicting all descriptors simultaneously) Deep Neural Network (DNN) architectures.
- Training Configuration: Train the models, likely using a framework like Python with deep learning libraries (e.g., PyTorch or TensorFlow). The training should employ data augmentation techniques, such as generating plausible tautomers, to improve model robustness and performance [80].
3. Model Validation:
- Performance Metrics: Quantify model accuracy using Root Mean Square Error (RMSE) for each solute descriptor (E, S, A, B, V, L) on a held-out test set.
- Benchmarking: Compare the performance of the novel DNN model against established prediction tools like QSPR from LSERD and ACD/Absolv.
- Application Testing: Evaluate the real-world predictive power by using the predicted descriptors in LSER equations to forecast well-known partition coefficients (e.g., Kow, Koa) and chromatographic retention data, then compare these predictions to experimental values [80].
Successful development and application of computational prediction models rely on a suite of data, software, and computational resources.
Table 2: Key Research Reagents and Resources for Descriptor Prediction
| Resource Name | Type | Function in Research | Access Information |
|---|---|---|---|
| UFZ-LSER Database [81] | Data | Primary source of experimentally derived Abraham solute descriptors for thousands of compounds. | Publicly available online (version 3.2.1) |
| Abraham Absolv Dataset [80] | Data | A widely used curated dataset of molecular structures and their solute descriptors for model training. | Described in literature; may require licensing for commercial use |
| ACD/Percepta (Absolv) [80] | Software | Commercial software providing QSPR predictions of solute descriptors; used as a performance benchmark. | Commercial license |
| ChemLLaMA / AbraLlama [81] | Software / Model | Fine-tuned Large Language Model for predicting solute descriptors and solvent parameters directly from SMILES strings. | Available on Hugging Face |
| Python with PyTorch/TensorFlow | Software | Core programming languages and deep learning libraries for building, training, and validating custom DNN models. | Open source |
| BigSolDB [82] | Data | A large experimental solubility dataset used for training and validating related property prediction models like fastsolv. | Publicly available |
The computational prediction of E and L descriptors has evolved decisively from traditional group-contribution methods to sophisticated, data-driven machine learning models. Deep Neural Networks and fine-tuned Large Language Models now offer highly accurate and complementary tools that overcome previous limitations with complex molecular structures [80] [81]. The integration of these advanced computational methods into the researcher's toolkit is empowering more reliable and expansive predictions of critical physicochemical properties, thereby accelerating innovation in drug discovery and environmental risk assessment. Future progress will likely hinge on the continued curation of high-quality experimental data and the development of even more interpretable and efficient machine-learning architectures.
Linear Solvation Energy Relationship (LSER) models represent a powerful quantitative approach for predicting solute partitioning and solvation properties across diverse chemical and biological systems. These models operate on the fundamental principle that free-energy-related properties of a solute can be correlated with its molecular descriptors through linear relationships [1]. The standard LSER model for solute transfer between two condensed phases takes the form: log(P) = cₚ + eₚE + sₚS + aₚA + bₚB + vₚVₓ where the capital letters represent solute-specific molecular descriptors and the lowercase letters represent complementary solvent-specific system coefficients [1]. The six fundamental Abraham solute descriptors include: McGowan's characteristic volume (Vₓ), the excess molar refraction (E), the dipolarity/polarizability (S), the hydrogen bond acidity (A), the hydrogen bond basicity (B), and the gas-liquid partition coefficient in n-hexadecane at 298 K (L) [1] [41].
Despite their widespread success in predicting partition coefficients, solubility, and chromatographic retention, LSER models frequently encounter challenges when outliers disrupt their predictive accuracy. These outliers often signal underlying issues with descriptor determination, model misspecification, or the presence of unique molecular interactions not adequately captured by the standard descriptor set. The identification and correction of such outliers is not merely a statistical exercise but a fundamental process for enhancing model robustness and expanding its applicability domains. This technical guide provides researchers with comprehensive methodologies for diagnosing, understanding, and correcting outlier-related issues in LSER applications, with particular emphasis on pharmaceutical and chemical development contexts where model reliability is paramount.
Understanding LSER Descriptors: Foundations and Limitations
The Six Fundamental Solute Descriptors
A precise understanding of each LSER descriptor's physical significance and determination method is essential for identifying potential sources of error and outlier behavior. The following table summarizes the core set of solute descriptors and their molecular interpretations:
Table 1: Fundamental LSER Solute Descriptors and Their Molecular Significance
| Descriptor | Molecular Interpretation | Determination Methods |
|---|---|---|
| Vₓ | McGowan's characteristic molecular volume | Calculated from molecular structure and atomic contributions |
| L | Gas-liquid partition coefficient in n-hexadecane at 298 K | Experimental measurement via gas chromatography |
| E | Excess molar refraction | Derived from refractive index measurements |
| S | Dipolarity/polarizability | Determined from solvatochromic comparison methods |
| A | Hydrogen bond acidity | Measured via solvation in hydrogen bond accepting phases |
| B | Hydrogen bond basicity | Measured via solvation in hydrogen bond donating phases |
Each descriptor quantifies a specific aspect of a molecule's interaction potential, with Vₓ representing dispersion forces, E capturing polarizability due to π- or n-electrons, S characterizing dipole-dipole and dipole-induced dipole interactions, and A and B quantifying hydrogen-bonding capabilities [1] [41]. The L descriptor incorporates multiple interaction types within a n-hexadecane reference system.
Thermodynamic Basis and Linearity Assumptions
The remarkable linearity of LSER models, even for strong specific interactions like hydrogen bonding, has a thermodynamic foundation that combines equation-of-state solvation thermodynamics with the statistical thermodynamics of hydrogen bonding [1]. This linearity persists because the free energy contributions of different interaction types are approximately additive, with each descriptor representing a distinct interaction mode. However, this additivity assumption can break down for molecules exhibiting significant conformational flexibility, intramolecular interactions, or unique electronic properties not adequately captured by the standard descriptor set [41]. Such breakdowns often manifest as outliers in LSER correlations and signal the need for descriptor refinement or model expansion.
Diagnostic Methodologies for Outlier Detection
Statistical Detection Methods
The first step in addressing outliers involves their systematic identification through statistical measures. Researchers should employ multiple diagnostic approaches to distinguish true outliers from naturally occurring variance:
- Residual Analysis: Examine the difference between observed and predicted values. Data points with standardized residuals exceeding ±2.5-3.0 standard deviations warrant further investigation.
- Leverage and Influence Measures: Calculate Hat values to identify observations with unusual predictor combinations (high leverage) and Cook's Distance to detect observations that exert disproportionate influence on model parameters.
- Cross-Validation Residuals: Implement leave-one-out or k-fold cross-validation to identify observations that the model consistently fails to predict accurately when excluded from the training set.
For complex datasets, consider employing robust regression methods that automatically downweight influential points, then compare the results with ordinary least squares to identify discrepant observations.
Thermodynamic Consistency Checking
Beyond statistical measures, outliers should be evaluated for thermodynamic consistency using known relationships between free energy, enthalpy, and entropy. The LSER model extends to solvation enthalpies through the relationship: ΔHₛ = cH + eHE + sHS + aHA + bHB + lHL [1]. Suspect observations that violate fundamental thermodynamic principles or exhibit significant deviations from expected enthalpy-entropy compensation patterns may indicate erroneous descriptor assignments or unaccounted molecular interactions.
Table 2: Common Outlier Types and Diagnostic Indicators in LSER Models
| Outlier Type | Statistical Signature | Potential Molecular Causes |
|---|---|---|
| Descriptor Error Outliers | Large residual for specific compounds across multiple systems | Incorrect A/B values for strong hydrogen bonders; miscalculated Vₓ for complex structures |
| Missing Interaction Outliers | Systematic residuals for compound classes | Missing ionization terms; specific halogen bonding; unique π-π interactions |
| Conformational Outliers | Inconsistent behavior across similar solvents | Conformation-dependent descriptor changes; intramolecular H-bonding |
| Ionization Outliers | Large errors for ionizable compounds at specific pH | Unaccounted ionization state; inadequate D⁺/D⁻ descriptor implementation |
Diagnostic Experimental Protocols
When statistical and thermodynamic diagnostics identify potential outliers, targeted experimental protocols can verify descriptor accuracy:
- Protocol for Suspect Hydrogen-Bonding Descriptors: For compounds with potentially misassigned A or B values, measure partition coefficients in at least three reference systems with characterized hydrogen-bonding properties (e.g., alkanes, alcohols, and ethers). Compare the observed hydrogen-bonding contribution with descriptor-predicted values.
- Protocol for Ionizable Compounds: For molecules with ionizable groups, measure partition coefficients or retention factors across a pH range that brackets the pKa. Plot logP against pH to identify the ionization profile and calculate the appropriate D⁺ or D⁻ descriptor [83].
- Protocol for Volumetric Descriptor Verification: For complex molecules with potentially misassigned Vₓ values, compare calculated McGowan volumes with experimental partial molar volume measurements in inert solvents.
Corrective Strategies and Model Refinement
Descriptor Expansion for Specific Interactions
When outliers result from molecular interactions not captured by the standard descriptor set, descriptor expansion offers a powerful corrective strategy. A modified LSER approach that includes separate ionization terms for acidic and basic solutes has demonstrated significant improvement in model performance, with reported R² values increasing from 0.846 to 0.987 and standard error decreasing from 0.163 to 0.051 for a butylimidazolium-based HPLC stationary phase [83]. The expanded model incorporates D⁺ and D⁻ descriptors to separately account for the ionization of basic and acidic solutes, respectively, providing more physically meaningful parameterization for ionizable compounds.
For compounds exhibiting specific halogen bonding or unique π-interactions, consider developing specialized descriptors that quantify these interaction potentials based on quantum chemical calculations of molecular surface properties or experimental measurements in carefully selected reference systems.
Quantum Chemical Approaches to Descriptor Determination
Recent advances integrate quantum chemical calculations with LSER frameworks to address descriptor assignment challenges, particularly for novel compounds lacking experimental data. Quantum Chemical LSER (QC-LSER) approaches derive molecular descriptors from COSMO-type quantum chemical calculations of molecular surface charge distributions [41]. This methodology offers several advantages for addressing outliers:
- Thermodynamic Consistency: QC-LSER provides a more thermodynamically consistent framework for handling self-solvation and strong specific interactions where traditional LSER assignments may fail [41].
- Conformational Sensitivity: The approach can account for conformation-dependent descriptor changes by calculating descriptors for different low-energy conformers and weighting them appropriately.
- Novel Compound Characterization: For newly synthesized compounds without experimental descriptor determinations, QC-LSER provides theoretically derived descriptors that can be validated through limited experimental measurements.
The workflow for implementing QC-LSER involves: (1) conducting conformational analysis to identify low-energy conformers; (2) performing COSMO calculations for each conformer; (3) generating sigma-profiles (molecular surface charge distributions); (4) calculating descriptors from the sigma-profiles; (5) validating against any available experimental data; and (6) iterating the calculations if discrepancies exceed acceptable thresholds.
Implementation Workflow for Outlier Management
The following diagram illustrates a comprehensive workflow for diagnosing and correcting outliers in LSER applications:
Diagram 1: LSER Outlier Diagnosis and Correction Workflow
Case Studies and Experimental Validation
Ionizable Compound Analysis with Expanded Descriptors
A compelling case study demonstrating the value of descriptor expansion involves the application of a modified LSER model to a butylimidazolium-based HPLC stationary phase. Initial modeling using only the standard six descriptors produced mediocre correlation (R² = 0.846) with substantial standard error (0.163). After incorporating separate D⁺ and D⁻ descriptors to account for ionization of basic and acidic solutes respectively, the model performance improved dramatically (R² = 0.987, SE = 0.051) [83]. This approach correctly predicted elution orders for ionizable analytes that deviated significantly from standard model predictions, resolving previous outlier behavior.
The experimental protocol for this approach involves:
- Mobile Phase Preparation: Prepare mobile phases with controlled pH using buffer systems that bracket the pKa values of target analytes.
- Retention Measurement: Measure retention factors for both ionizable and neutral reference compounds across the pH range.
- Descriptor Determination: Calculate D⁺ for basic compounds as logD - logP at pH values where the compound is fully ionized, and similarly D⁻ for acidic compounds.
- Model Implementation: Incorporate the ionization descriptors into the expanded LSER model: log(k) = c + eE + sS + aA + bB + vVₓ + d⁺D⁺ + d⁻D⁻.
- Validation: Test the expanded model against a separate validation set of ionizable compounds.
Quantum Chemical Descriptor Determination
For compounds where experimental descriptor determination is challenging, quantum chemical approaches offer an alternative pathway. A recent study demonstrated the determination of solute descriptors for 13 new compounds using reversed-phase liquid chromatography with binary and ternary solvent systems on a single stationary phase [84]. The approach successfully replicated descriptor values from the established WSU descriptor database for 31 reference compounds, with standard errors for estimated descriptors ranging from 0.019 to 0.080, demonstrating the method's precision.
The experimental protocol involves:
- Stationary Phase Selection: Choose a stationary phase with well-characterized LSER system parameters.
- Solvent System Design: Employ binary and ternary solvent mixtures (e.g., water-methanol, water-acetonitrile, and water-methanol-acetonitrile) to create diverse solvation environments.
- Retention Measurement: Measure retention factors for both reference compounds with known descriptors and new target compounds.
- Descriptor Calculation: Use multi-linear regression against the system parameters to calculate unknown descriptors for target compounds.
- Cross-Validation: Verify descriptor accuracy by predicting retention in different solvent systems and comparing with experimental values.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Materials for LSER outlier Investigation and Method Development
| Reagent/Material | Specification | Application Function |
|---|---|---|
| n-Hexadecane | HPLC grade, ≥99% | Reference solvent for L descriptor determination |
| Reference Solvent Set | Alkanes, alcohols, ethers, ketones | Characterizing system parameters for new stationary phases |
| Buffer Systems | pH 3-10, volatile buffers preferred | Mobile phase modification for ionizable compounds |
| Characterized Stationary Phases | C18, phenyl, HILIC, ion-exchange | Multi-system retention measurement for descriptor determination |
| Quantum Chemical Software | COSMO-RS, Gaussian, ORCA | Calculation of molecular descriptors from first principles |
| LC-MS System | High precision UHPLC with MS detection | Accurate retention factor measurement for diverse compounds |
The effective management of outliers in LSER applications requires a systematic approach that combines statistical diagnostics with molecular-level understanding of interaction mechanisms. Rather than treating outliers as statistical nuisances to be excluded, researchers should view them as opportunities to identify limitations in current descriptor sets and modeling approaches. The integration of expanded descriptor sets for specific interactions like ionization, coupled with quantum chemically derived descriptors for novel compounds, provides a powerful framework for enhancing model robustness and expanding applicability domains. As LSER methodologies continue to evolve, particularly through integration with quantum chemical calculations and machine learning approaches, the systematic diagnosis and correction of outlier behavior will remain essential for advancing predictive capability in pharmaceutical development, environmental chemistry, and materials design.
In the field of quantitative structure-activity relationship (QSAR) modeling, particularly research involving Linear Solvation Energy Relationship (LSER) solute descriptors, the robustness of predictive models is paramount. Descriptor-based models, which translate molecular structures into predictive attributes for biological activity or physicochemical properties, are inherently susceptible to overfitting and optimistic performance estimates. This technical guide examines the critical role of cross-validation techniques in mitigating these risks, providing researchers and drug development professionals with rigorous methodologies for model assessment. By implementing systematic validation protocols, scientists can ensure their LSER-based models deliver reliable, generalizable predictions that advance drug discovery and molecular design.
Descriptor-based models represent chemical structures numerically, enabling the prediction of complex properties from simplified parameters. Within this domain, Linear Solvation Energy Relationship (LSER) solute descriptors provide a powerful framework for understanding and predicting how molecules interact with their environment. These descriptors quantify key solvation characteristics, typically encapsulated in parameters such as Vx (characteristic molecular volume), E (excess molar refractivity), S (dipolarity/polarizability), A (hydrogen-bond acidity), B (hydrogen-bond basicity), and L (gas-liquid partition coefficient on hexadecane). The fundamental challenge in developing such models lies in their inherent complexity and the risk of capturing dataset-specific noise rather than generalizable relationships.
The core vulnerability of these models is overfitting, where a model performs well on its training data but fails to predict unseen samples accurately. This occurs when models become excessively complex, tailoring themselves to idiosyncrasies in the training set. Without proper validation, such models yield misleadingly optimistic performance estimates, potentially derailing research directions or drug development programs based on flawed predictions. Cross-validation addresses this fundamental issue by providing a more realistic assessment of how models will perform on new, unseen data [85] [86].
The statistical learning foundation of cross-validation is well-established, with early developments dating to Quenouille (1949) and Stone (1974) [87]. These methods have evolved into essential tools for modern computational chemistry and drug discovery, where model reliability directly impacts research validity and resource allocation.
Core Cross-Validation Techniques: Methodologies and Applications
Cross-validation techniques operate through a common principle: repeatedly partitioning available data into training and validation subsets to simulate performance on unseen data. The following sections detail the primary methodologies, their implementation protocols, and specific applications to descriptor-based modeling.
Hold-Out Cross-Validation
Experimental Protocol:
- Random Partitioning: Split the dataset D of size N into two mutually exclusive sets: a training set (Dtrain) and a test set (Dtest). Common split ratios are 70:30 or 80:20, training to test.
- Model Training: Train the predictive model (e.g., Multiple Linear Regression, Support Vector Machine) using only the data in D_train.
- Performance Evaluation: Apply the trained model to D_test and calculate relevant performance metrics (e.g., R², Mean Squared Error, Root Mean Squared Error).
- Implementation: The partition should be performed using a stratified approach if the dataset contains class imbalances for classification problems, or through simple random sampling for regression tasks like those common with LSER descriptors.
Table 1: Hold-Out Cross-Validation Profile
| Characteristic | Specification |
|---|---|
| Typical Split Ratio | 70% Training / 30% Test or 80% Training / 20% Test |
| Computational Cost | Low |
| Data Utilization | Partial (Uses only a portion of data for training) |
| Variance of Estimate | High (Highly dependent on a single random split) |
| Primary Use Case | Initial model prototyping, very large datasets |
While straightforward to implement, Hold-Out validation's major limitation is that its evaluation can be highly dependent on a single, arbitrary split of the data, potentially leading to unstable performance estimates [85].
k-Fold Cross-Validation
Experimental Protocol:
- Dataset Division: Randomly shuffle the dataset D and partition it into k approximately equal-sized folds (F₁, F₂, ..., Fₖ). Common values for k are 5 or 10.
- Iterative Training and Validation: For each iteration i (where i = 1 to k):
- Designate fold Fᵢ as the validation set.
- Combine the remaining k-1 folds to form the training set.
- Train the model on the combined training set.
- Validate the trained model on Fᵢ and store the performance metric.
- Performance Aggregation: Calculate the final model performance by averaging the performance metrics from all k iterations. This provides a more robust estimate than a single Hold-Out test.
Table 2: k-Fold Cross-Validation Profile
| Characteristic | Specification |
|---|---|
| Common k Values | 5, 10 |
| Computational Cost | Moderate (Model is trained k times) |
| Data Utilization | Complete (Each data point is used for validation once) |
| Variance of Estimate | Moderate (Lower than Hold-Out) |
| Primary Use Case | Standard model evaluation and hyperparameter tuning |
k-Fold Cross-Validation provides a better trade-off between bias and variance than the Hold-Out method. It uses all data for both training and validation, leading to a more reliable performance estimate that is less dependent on a single data split [85].
Leave-One-Out Cross-Validation (LOOCV)
Experimental Protocol:
- Iteration Setup: For a dataset D with N samples, configure N iterations.
- Single-Sample Validation: For each iteration i (where i = 1 to N):
- Select the i-th sample as the validation set.
- Use the remaining N-1 samples as the training set.
- Train the model on the N-1 samples.
- Compute the prediction error for the single validation sample.
- Final Calculation: The overall performance metric (e.g., Mean Squared Error) is computed as the average of the prediction errors from all N iterations.
LOOCV is a special case of k-Fold Cross-Validation where k equals the number of samples (N). It is particularly useful for very small datasets, as it maximizes the training data used in each iteration. However, it is computationally expensive for large N and can yield estimates with high variance, as the model is tested on a single data point each time [85].
Stratified and Repeated k-Fold Cross-Validation
For classification problems with imbalanced class distributions, Stratified k-Fold Cross-Validation is essential. This technique ensures that each fold maintains the same proportion of class labels as the complete dataset, preventing folds with missing classes which would lead to biased performance estimates [85].
Repeated k-Fold Cross-Validation involves performing the standard k-Fold process multiple times (e.g., 10 repetitions of 5-Fold) with different random partitions of the data. The final performance is averaged over all runs and all folds. This further reduces the variability of the performance estimate and provides a more robust assessment of model performance, though it increases computational cost significantly [85].
Implementation in LSER Descriptor Research
The application of cross-validation within LSER research demands specific considerations due to the nature of the descriptors and their applications in predictive toxicology and drug development.
Integration with Model Development Workflows
Inverse least-squares modeling, commonly employed with LSER descriptors, benefits significantly from cross-validation to prevent overfitting to the specific vapor or solute dataset [88]. The workflow typically follows this sequence: (1) descriptor calculation and curation, (2) model design and feature selection, (3) cross-validation setup, (4) model training within the cross-validation loop, and (5) final model assessment and interpretation.
For LSER models predicting serious adverse drug reactions like Torsade de Pointes (TdP), robust validation is critical. Yap et al. successfully used Support Vector Machines (SVM) with LSER descriptors and leave-one-out cross-validation to predict TdP-causing potential, achieving high prediction accuracies (97.4% for TdP-causing agents and 84.6% for non-TdP-causing agents) [89]. This demonstrates the power of combining appropriate machine learning algorithms with rigorous validation in a pharmacological context.
Addressing Specialized Challenges
LSER descriptor research presents unique validation challenges that standard protocols must adapt to handle:
- Descriptor Collinearity: LSER parameters (Vx, E, S, A, B, L) can exhibit correlations. Cross-validation must be coupled with feature selection or regularization techniques (e.g., Ridge Regression, LASSO) within each training fold to avoid model instability.
- Limited Dataset Sizes: Many physicochemical and biological studies have relatively small sample sizes (N < 100). In such cases, LOOCV or repeated k-Fold validation provides the most reliable error estimates without sacrificing training data.
- Domain Applicability: Models must be validated to ensure they perform well across the entire chemical space of interest, not just the specific compounds used in training. This may require stratified splits based on chemical scaffold or functional groups.
Performance Assessment and Quality Measures
The accurate interpretation of cross-validation results is as critical as its proper execution. Robust quality measures are essential for comparing models and assessing their predictive confidence.
Core Performance Metrics
The choice of performance metric should align with the research objective and the nature of the dependent variable. For continuous outcomes typical in LSER modeling (e.g., partition coefficients, solubility, retention times), Mean Squared Error (MSE) and Root Mean Squared Error (RMSE) are most common, as they penalize large errors more heavily. The Coefficient of Determination (R²) indicates the proportion of variance explained by the model, providing an intuitive measure of goodness-of-fit.
For classification tasks (e.g., toxic vs. non-toxic), metrics such as accuracy, precision, recall, F1-score, and AUC-ROC are appropriate. The confusion matrix serves as the foundation for these metrics.
Estimating Robustness and Confidence
Recent research emphasizes the importance of quantifying the robustness of the cross-validation estimates themselves. Most et al. (2024) investigated quality measures derived from cross-validation, highlighting the value of reporting confidence bounds for performance metrics [86]. These bounds can be estimated via:
- Standard Deviation Across Folds: Calculating the mean and standard deviation of a metric (e.g., R²) across all k-folds provides insight into the stability of the model performance.
- Bootstrapping: Resampling the cross-validation results can generate empirical confidence intervals for performance metrics.
- Repeated Cross-Validation: Performing multiple runs of k-Fold CV with different random seeds and analyzing the distribution of results offers the most comprehensive view of model robustness.
Table 3: Cross-Validation Technique Selection Guide
| Criterion | Recommended Technique | Rationale |
|---|---|---|
| Large Dataset (N > 10,000) | Hold-Out | Single split is sufficient and computationally efficient. |
| Standard Dataset Size | k-Fold (k=5 or 10) | Optimal balance of bias, variance, and computational cost. |
| Very Small Dataset (N < 100) | Leave-One-Out (LOOCV) | Maximizes training data in each iteration; low bias. |
| Imbalanced Classification | Stratified k-Fold | Preserves class distribution in each fold; prevents bias. |
| Highest Robustness Requirement | Repeated k-Fold | Reduces variability of the performance estimate. |
The Scientist's Toolkit: Research Reagent Solutions
Implementing robust cross-validation requires both conceptual understanding and practical tools. The following table outlines key "research reagents" — software libraries and computational resources — essential for applying these techniques to LSER descriptor research.
Table 4: Essential Research Reagent Solutions for Cross-Validation
| Tool/Resource | Function | Application in LSER Research |
|---|---|---|
| scikit-learn (Python) | Comprehensive ML library | Provides ready-to-use implementations of all major CV techniques (e.g., KFold, LeaveOneOut, cross_val_score). |
| R Statistical Language | Statistical computing and graphics | Offers extensive CV capabilities via packages like caret, mlr, and boot for model training and validation. |
| Molecular Descriptor Software | Descriptor calculation | Tools like RDKit, PaDEL, or Dragon generate the initial LSER-like descriptors (Vx, E, S, A, B, L) from molecular structures. |
| High-Performance Computing (HPC) Cluster | Parallel processing | Accelerates computationally intensive tasks like Repeated k-Fold CV or LOOCV on large molecular datasets. |
| Jupyter Notebook / RStudio | Interactive development | Facilitates iterative model development, visualization, and documentation of the entire CV workflow. |
Cross-validation is not merely a supplementary step but a foundational component of rigorous descriptor-based predictive modeling. For researchers working with LSER solute descriptors, adopting these techniques is essential for developing models that truly generalize beyond their training data. The choice of a specific cross-validation strategy—whether k-Fold for standard datasets, LOOCV for limited samples, or stratified approaches for imbalanced data—directly impacts the reliability of performance estimates and the validity of scientific conclusions. As machine learning continues to transform computational chemistry and drug development, the disciplined application of cross-validation will remain the key differentiator between speculative models and robust, trustworthy predictive tools that can confidently guide decision-making in research and development.
Linear Solvation Energy Relationship (LSER) models are powerful tools used across chemical, environmental, and pharmaceutical sciences to predict how molecules partition between different phases or interact with biological and environmental systems. The standard Abraham LSER model describes these interactions using a set of five core molecular descriptors (Vx, E, S, A, B) with an alternative model sometimes using L instead of Vx [72]. These descriptors quantitatively represent specific molecular properties: Vx (McGowan's characteristic volume in cm³ mol⁻¹/100) characterizes the cavity formation energy; E represents the excess molar refraction; S indicates dipolarity/polarizability; A quantifies hydrogen-bond donor acidity; and B represents hydrogen-bond acceptor basicity [90] [91]. The alternative descriptor L defines the gas-liquid partition coefficient in n-hexadecane at 298 K [90].
Sensitivity analysis within the LSER framework systematically evaluates how changes in these molecular descriptors influence the output property of interest (e.g., partition coefficient, retention factor, or permeability). This analysis is crucial for researchers to identify which molecular properties dominantly control a specific process, enabling more efficient experimental design, compound optimization, and predictive modeling. For drug development professionals, understanding descriptor sensitivity allows for rational optimization of key properties like intestinal absorption, blood-brain barrier penetration, and solubility [91].
Foundational Principles of LSER Models
The foundational LSER model expresses a free energy-related property as a linear combination of solute descriptors and system-specific coefficients. The general form for a partitioning process between two condensed phases is:
log SP = c + eE + sS + aA + bB + vVx
Where SP is the solute property of interest (e.g., partition coefficient, retention factor), the uppercase letters (E, S, A, B, Vx) are the solute-specific molecular descriptors, and the lowercase letters (c, e, s, a, b, v) are system-specific coefficients determined through multiple linear regression of experimental data [72]. These system coefficients reflect the complementary properties of the phases involved and indicate how much a specific interaction contributes to the overall solvation or partitioning process.
For processes involving gas-to-condensed phase transfer, the model often uses the L descriptor instead of Vx:
log K = c + eE + sS + aA + bB + lL
The model's predictive power relies heavily on the accurate determination of both solute descriptors and system coefficients. The solute descriptors are considered intrinsic molecular properties that are, in principle, transferable between different systems, while the system coefficients are specific to the particular phases or conditions being studied [90]. This separation between molecular and system properties forms the basis for sensitivity analysis, as it allows researchers to determine how modifying specific molecular features (changing descriptors) will affect the property of interest in a given system.
Methodologies for Sensitivity Analysis
Statistical Approaches for Determining Descriptor Influence
Multiple linear regression serves as the primary statistical method for determining system coefficients in LSER models. The magnitude and sign of the resulting coefficients (v, e, s, a, b) directly indicate each descriptor's influence on the property being modeled. A larger absolute value of a system coefficient signifies that the corresponding molecular descriptor has a greater impact on the property in that specific system [72]. For example, in reversed-phase liquid chromatography, the v coefficient (multiplying Vx) is typically positive and significant, indicating that cavity formation strongly influences retention, while in normal-phase systems, s and a coefficients often dominate.
The standard error of the regression coefficients provides crucial information about the reliability of sensitivity conclusions. As noted in recent research, "the standard error of the estimator depends on both the number of data points and the range covered by the descriptor values" [72]. This relationship can be expressed as SE ∝ 1/(σₓ√n), where σₓ is the standard deviation of the descriptor values and n is the number of solutes. This statistical foundation underscores why selecting solutes with descriptor values that span a wide range is more important than simply using a large number of solutes when constructing LSER models for sensitivity analysis.
Multicollinearity presents a significant challenge in sensitivity analysis, as correlated descriptors make it difficult to isolate individual contributions. The Pearson correlation coefficient and Average Absolute Correlation (AAC) metric quantify descriptor interdependence [72]. Strategy 1 for solute selection focuses on minimizing AAC to reduce multicollinearity, while Strategy 2 prioritizes maximizing descriptor range to improve coefficient estimation reliability, with research showing Strategy 2 often provides better alignment with ground truth values [72].
Experimental Design for Robust Sensitivity Analysis
Selecting an appropriate solute set forms the foundation of reliable sensitivity analysis. Research demonstrates that choosing solutes with maximum differences between normalized descriptors (Strategy 2) creates a chemically diverse set that spans the chemical space of interest, leading to more accurate determination of system coefficients compared to approaches focused solely on minimizing descriptor correlation [72]. The minimal number of solutes required is theoretically six (to determine the six system coefficients), but in practice, using 20-50 carefully selected solutes significantly improves reliability.
Monte Carlo simulations with added random normal noise during multiple linear regression help assess the robustness of sensitivity conclusions. As noted in recent studies, performing "10,000 iterations which checks for different combinations of 20 and 50 compounds... helps to analyse how noise impacts the coefficient distributions" [72]. These simulations generate Gaussian-shaped coefficient distributions due to the central limit theorem, with narrower distributions indicating more reliable sensitivity rankings.
For ionizable compounds, the standard LSER model requires modification to account for pH-dependent speciation. The D solute descriptor represents the degree of ionization at the mobile phase pH and can be separated into D+ and D- components for basic and acidic solutes respectively [92]. The modified LSER equation becomes: log k = c + eE + sS + aA + bB + vV + d₊D₊ + d₋D₋. The relative magnitudes of d₊ and d₋ coefficients then indicate how ionization affects the property for acids versus bases in the system.
Advanced Descriptor Modification Techniques
Descriptor scaling and modification can enhance sensitivity analysis for specific applications. Scaled Polar Surface Area (PSA) descriptors address limitations of standard PSA by accounting for varying hydrogen-bond strengths of different functional groups [91]. For example, scaling factors can be applied to N-H and O-H groups based on their known hydrogen bond donor strengths, which vary significantly (e.g., dimethylamine at 0.08 versus tetrazole at 0.79 on Abraham's free energy scale) [91].
Splitting composite descriptors into component parts provides more detailed sensitivity information. Partitioned Total Surface Area (PTSA) deconvolutes PSA and molecular surface area into separate descriptors, offering marked improvements over traditional PSA methods for modeling intestinal absorption of drugs [91]. Similarly, separating hydrogen-bonding descriptors into distinct acid and base components allows more precise determination of how each hydrogen-bonding type influences the property.
Quantum chemical calculations enable the development of novel descriptors like the QC-LSER descriptors based on molecular surface charge densities (σ-profiles) [90]. These calculated descriptors can predict hydrogen-bonding interaction free energies using the relationship -ΔG₁₂ʰᵇ = 5.71(α₁β₂ + β₁α₂) kJ/mol at 25°C, where α and β are effective HB acidity and basicity descriptors [90]. This approach provides a priori sensitivity predictions without extensive experimental data.
Application-Specific Descriptor Sensitivity
Chromatographic Systems
In reversed-phase liquid chromatography, the Vx descriptor typically exhibits the highest sensitivity, reflected by the large positive v system coefficient, indicating that cavity formation (solvophobic effect) dominates retention. For example, in a butylimidazolium-based stationary phase, retention properties were found to be similar to phenyl phases, with the v coefficient being particularly significant [92]. The b coefficient (multiplying B) is generally negative in reversed-phase systems, reflecting competition for hydrogen-bonding sites between solutes and the aqueous mobile phase.
Supercritical fluid chromatography (SFC) with polar stationary phases shows markedly different sensitivity patterns. Studies of over 200 drug-like compounds found that "the dominant contribution to positive retention was the hydrogen bond donor acidity of the solutes" represented by the A descriptor, particularly for pyridine and amino columns [93]. The relative sensitivity ranking in SFC often follows A > B > S > Vx > E, contrasting sharply with reversed-phase LC where Vx dominates.
For ionizable compounds in chromatographic systems, the ionization descriptors (D+ and D-) can become the most sensitive parameters. Research shows that "ionization of weakly acidic analytes should lead to an increase in retention whereas ionization of weakly basic compounds should lead to a reduction in retention" on a butylimidazolium stationary phase [92]. This produces positive d₋ coefficients for acids and negative d₊ coefficients for bases, with the magnitude of these coefficients determining the sensitivity to ionization state.
Pharmaceutical and Biological Partitioning
In blood-brain barrier partitioning, the A and B descriptors relating to hydrogen-bonding typically show the highest sensitivity, with increased hydrogen-bonding capacity reducing penetration. Research indicates that "PSA has been used, either alone or in combination with other descriptors such as log Poct, to model a wide range of biological properties such as blood-brain distribution" [91]. The relative sensitivity often follows A ≈ B > S > Vx, reflecting the importance of both hydrogen-bond donor and acceptor properties in biological membrane penetration.
Intestinal absorption displays a well-defined sensitivity threshold related to Polar Surface Area (PSA). Studies show that "molecules with a PSA ≤ 60 Ų will exhibit high and almost complete intestinal absorbance, while molecules with a PSA ≥ 140 Ų exhibit poor intestinal absorbance" [91]. Since PSA correlates with hydrogen-bonding descriptors A and B, these descriptors show high sensitivity in absorption models, with the A descriptor often being more influential than B.
Table 1: Relative Descriptor Sensitivity Across Application Domains
| Application Domain | Sensitivity Ranking (High to Low) | Dominant Physical Interaction |
|---|---|---|
| Reversed-Phase Chromatography | Vx > B > A ≈ S > E | Cavity formation/Solvophobic effect |
| Supercritical Fluid Chromatography | A > B > S > Vx > E | Hydrogen-bond donation |
| Blood-Brain Barrier Penetration | A ≈ B > S > Vx > E | Hydrogen-bonding capacity |
| Octanol-Water Partitioning | Vx > A ≈ B > S > E | Cavity formation & Hydrogen-bonding |
| Intestinal Absorption | A > B > Vx > S > E | Hydrogen-bond donation & Size |
Environmental Partitioning Systems
In environmental systems like soil-water partitioning, the Vx descriptor generally shows the highest sensitivity, reflected by large positive v coefficients, indicating the dominance of hydrophobic interactions. The LSER model for soil-water partitioning coefficients typically takes the form log Kₛw = c + vVx + bB + aA + ..., with the v coefficient being substantially larger than other coefficients for most natural organic sorbents.
Air-to-condensed phase partitioning exhibits distinct sensitivity patterns where the L descriptor replaces Vx in the model. For air-organic phase partitioning, the l coefficient (multiplying L) typically shows the highest sensitivity, followed by the a and b coefficients for hydrogen-bonding. The sensitivity ranking generally follows L > A ≈ B > S > E, reflecting the importance of dispersion interactions and hydrogen-bonding in these systems.
Table 2: Experimental Protocols for Descriptor Sensitivity Determination
| Protocol Step | Methodological Details | Critical Parameters |
|---|---|---|
| Solute Set Selection | Select 20-50 solutes using maximum dissimilarity strategy; normalize descriptors (0-1) then maximize Euclidean distances [72] | Average Absolute Correlation < 0.6; wide descriptor range coverage |
| Data Measurement | Measure partition/retention factors in triplicate; include internal standards; control temperature (±0.1°C) | Minimum R² > 0.98 for calibration curves; precise pH control (±0.02 units) |
| Regression Analysis | Multiple linear regression with variance inflation factor (VIF) check; VIF > 5 indicates multicollinearity issues [72] | Significance level p < 0.05; residual analysis for outliers |
| Sensitivity Validation | Monte Carlo simulations with 10,000 iterations adding random normal noise (σ = 0.05-0.2 log units) [72] | Coefficient distributions should be Gaussian with mean near ground truth |
| Model Application | Predict properties for test set compounds (20% of total) not used in model development | Predictive R² > 0.85; average absolute error < 0.3 log units |
Research Reagent Solutions for LSER Studies
Table 3: Essential Research Reagents and Materials for LSER Experiments
| Reagent/Material | Specifications | Application Function |
|---|---|---|
| Reference Solutes | 40-50 compounds with known descriptors spanning chemical space; purity >99% [72] | Provide calibration set for determining system coefficients through multiple linear regression |
| UFZ-LSER Database | Database containing >5,000 compounds with pre-calculated descriptors [94] | Reference source for solute descriptors; enables solute selection and descriptor verification |
| Chromatographic Columns | Butylimidazolium, C18, phenyl, cyano, amino, diol stationary phases [92] [93] | Provide varied interaction environments for probing specific descriptor sensitivities |
| Quantum Chemistry Software | TURBOMOLE, DMol3, MATERIALS STUDIO suite, or SCM suite [90] | Calculate σ-profiles and develop novel QC-LSER descriptors for advanced sensitivity analysis |
| Mobile Phase Modifiers | HPLC-grade methanol, acetonitrile, water; ammonium acetate, formic acid [92] [93] | Control solvent strength and pH; modify selectivity to enhance sensitivity to specific descriptors |
Sensitivity analysis of LSER descriptors provides crucial insights into the molecular interactions governing partitioning behavior across diverse chemical, pharmaceutical, and environmental systems. The relative influence of Vx, E, S, A, B, and L descriptors varies significantly depending on the specific application, with Vx dominating in reversed-phase chromatographic systems and hydrophobic partitioning, while A and B descriptors show heightened sensitivity in hydrogen-bonding dependent processes like biological membrane penetration and normal-phase separations. Robust sensitivity analysis requires careful experimental design, including selection of chemically diverse solute sets that maximize descriptor range while managing multicollinearity, followed by rigorous statistical validation using Monte Carlo methods. The continuing development of novel descriptors, including scaled PSA approaches and quantum chemically-derived parameters, promises to further enhance our ability to precisely quantify descriptor sensitivity for increasingly complex chemical systems.
Validating LSER Approaches: Comparative Analysis with Alternative Solvation Models
Benchmarking LSER Predictions Against Experimental Partition Coefficient Data
Linear Solvation Energy Relationships (LSERs) are powerful quantitative structure-activity relationship (QSAR) models that predict how a solute will distribute itself between two phases based on its molecular properties. The foundational LSER model for partitioning systems is described by the Abraham equation, which utilizes a set of solute descriptors to characterize specific interaction capabilities. For researchers in drug development, accurately predicting partition coefficients is crucial for understanding drug absorption, distribution, and leaching from packaging materials into pharmaceutical formulations.
The core LSER equation for partition coefficients between a polymer phase and water takes the general form:
log K = c + eE + sS + aA + bB + vV
Where the system parameters (c, e, s, a, b, v) are characteristics of the specific partitioning system and phases involved, and the solute descriptors (E, S, A, B, V, L) represent the following molecular properties of the compound of interest:
- V - McGowan's characteristic molecular volume in units of cm³ mol⁻¹/100
- E - Excess molar refractivity, which models polarizability contributions from n- and π-electrons
- S - Solute dipolarity/polarizability, representing the ability to engage in dipole-dipole and dipole-induced dipole interactions
- A - Solute overall hydrogen-bond acidity
- B - Solute overall hydrogen-bond basicity
- L - The logarithm of the gas-hexadecane partition coefficient at 298 K
This framework allows for the robust prediction of partition coefficients for any neutral compound with known descriptors, making it invaluable for pharmaceutical scientists modeling drug behavior and excipient compatibility.
Quantitative Benchmarking of LSER Model Performance
Core LSER Model for LDPE-Water Partitioning
Recent research has yielded a highly accurate LSER model for predicting partition coefficients between low-density polyethylene (LDPE) and water, a system particularly relevant to pharmaceutical packaging [95] [96]. The developed model is expressed as:
log K_{i,LDPE/W} = -0.529 + 1.098E - 1.557S - 2.991A - 4.617B + 3.886V
This model was established using experimental partition coefficients for 156 chemically diverse compounds, demonstrating exceptional accuracy and precision with a coefficient of determination (R²) of 0.991 and root mean square error (RMSE) of 0.264 [95] [96]. The high R² value indicates that the model explains over 99% of the variance in the experimental data, while the low RMSE suggests strong predictive capability.
Comprehensive Model Performance Metrics
Table 1: Performance metrics for the LDPE-water LSER model under different validation conditions
| Validation Type | Number of Compounds | R² Value | RMSE | Descriptor Source |
|---|---|---|---|---|
| Initial Calibration | 156 | 0.991 | 0.264 | Experimental |
| Independent Validation | 52 | 0.985 | 0.352 | Experimental |
| Predictive Scenario | 52 | 0.984 | 0.511 | QSPR-predicted |
The independent validation set, comprising approximately 33% of the total observations (n=52), confirmed the model's robustness when using experimental solute descriptors [95]. When Quantitative Structure-Property Relationship (QSPR)-predicted descriptors were employed instead of experimental ones—a common scenario for new compounds without extensive experimental characterization—the model maintained strong predictive power (R² = 0.984) with a modest increase in RMSE to 0.511 [95]. This benchmark is particularly valuable for drug development professionals working with novel chemical entities where experimental descriptors may not be available.
Table 2: Comparison of LSER model performance across different polymeric phases
| Polymer Phase | System Constant (c) | V Coefficient | B Coefficient | Key Interaction Characteristics |
|---|---|---|---|---|
| LDPE | -0.529 | 3.886 | -4.617 | High hydrophobicity dominance |
| LDPE (amorphous) | -0.079 | - | - | Similar to n-hexadecane/water |
| Polydimethylsiloxane (PDMS) | - | - | - | Comparable to LDPE for log K > 4 |
| Polyacrylate (PA) | - | - | - | Stronger sorption of polar compounds |
| Polyoxymethylene (POM) | - | - | - | Enhanced polar interactions |
The benchmarking analysis reveals that polymers with heteroatomic building blocks (PA, POM) exhibit stronger sorption than LDPE for more polar, non-hydrophobic compounds up to a log K_{i,LDPE/W} range of 3-4 [95]. Above this range, all four polymers demonstrate roughly similar sorption behavior, highlighting the domain of applicability for each material in pharmaceutical applications.
Experimental Protocols for LSER Validation
Partition Coefficient Determination Methodology
The experimental determination of partition coefficients between LDPE and water follows a rigorous protocol to ensure data quality and reproducibility. The general workflow involves:
Sample Preparation: LDPE films of standardized dimensions and thickness are pre-cleaned to remove potential contaminants. An aqueous solution containing the compound(s) of interest at known concentrations is prepared using high-purity water.
Equilibration Phase: The LDPE film is immersed in the aqueous solution and maintained at constant temperature (typically 25°C or 37°C for pharmaceutical applications) with continuous agitation for a predetermined period—usually 24-48 hours—to ensure equilibrium is reached.
Separation and Analysis: Following equilibration, the polymer film is removed from the aqueous phase and briefly rinsed to remove adhering solution. The concentration of the compound in both phases is quantified using appropriate analytical techniques such as high-performance liquid chromatography (HPLC), gas chromatography (GC), or mass spectrometry (MS).
Calculation: The partition coefficient is calculated as K = Cp / Cw, where Cp is the concentration in the polymer phase and Cw is the concentration in the water phase at equilibrium. The logarithm of this value is used for LSER modeling.
This methodology requires careful control of experimental conditions including temperature, pH, ionic strength, and absence of co-solvents that might influence partitioning behavior.
Solute Descriptor Determination Methods
Table 3: Experimental methods for determining LSER solute descriptors
| Descriptor | Primary Experimental Methods | Key Measurement Principle |
|---|---|---|
| V | Density measurement, molecular modeling | McGowan's characteristic volume based on molecular structure |
| E | Refractometry | Excess molar refractivity measured at 20°C using sodium D line |
| S | Chromatographic retention (HPLC, GC) | Solute dipolarity derived from retention behavior on stationary phases |
| A | Spectroscopic titration | Hydrogen-bond acidity measured through complexation with reference bases |
| B | Spectroscopic titration | Hydrogen-bond basicity measured through complexation with reference acids |
| L | Gas chromatography | Gas-hexadecane partition coefficient determined by GC retention |
For compounds where experimental determination of descriptors is impractical, QSPR tools provide predicted values, though with some compromise in accuracy as evidenced by the increased RMSE in validation studies [95].
LSER Benchmarking Workflow: This diagram illustrates the sequential process for developing and validating LSER models, from initial experimental design to final application of the benchmarked model.
Key Research Reagent Solutions and Materials
Table 4: Essential materials and resources for LSER partitioning studies
| Resource | Specification/Function | Application Context |
|---|---|---|
| LDPE Films | High-purity, standardized thickness (50-200 µm) | Primary polymer phase for partition coefficient determination |
| Reference Compounds | Chemically diverse set with known descriptors | Model calibration and validation |
| Chromatography Systems | HPLC/GC with various detection methods | Quantification of solute concentrations in both phases |
| UFZ-LSER Database | Web-based curated database of solute descriptors | Source of experimental descriptor values [97] |
| QSPR Prediction Tools | Software for predicting solute descriptors | Generating descriptors for novel compounds without experimental data |
| Constant Temperature Bath | Precision control (±0.1°C) | Maintaining consistent temperature during equilibration |
The UFZ-LSER database represents a particularly valuable resource, providing free access to a curated collection of solute descriptors and enabling the calculation of partition coefficients for any neutral compound with a known structure for a given two-phase system [97]. This database significantly streamlines the initial phases of LSER model development.
Advanced Implementation and Domain Considerations
Amorphous Polymer Phase Correction
When targeting more precise alignment with liquid-phase partitioning behavior, the partition coefficient can be converted to account for the amorphous fraction of the polymer as the effective phase volume. The modified LSER model for amorphous LDPE partitioning is expressed as:
log K_{i,LDPEamorph/W} = -0.079 + 1.098E - 1.557S - 2.991A - 4.617B + 3.886V
This adjustment, which primarily affects the system constant (changed from -0.529 to -0.079), renders the model more similar to a corresponding LSER model for n-hexadecane/water partitioning [95]. This refinement is particularly relevant for pharmaceutical scientists interested in biomimetic partitioning that more closely resembles biological membrane transport.
Domain of Applicability and Limitations
The predictability of LSER models strongly correlates with both the quality of experimental partition coefficients and the chemical diversity of the training set [95]. Models developed with limited chemical diversity in the training compounds may demonstrate reduced predictive accuracy for structurally novel compounds outside this domain.
Current LSER models are primarily validated for neutral compounds, and their application to ionizable pharmaceuticals requires additional considerations for the ionic species. Furthermore, the models assume no specific interactions between solutes beyond the parameterized descriptors, which may limit accuracy for compounds with unusual structural features or strong, specific intermolecular interactions.
LSER Variable Relationships: This diagram illustrates how the six fundamental solute descriptors contribute to the prediction of partition coefficients in LSER models.
The comprehensive benchmarking of LSER predictions against experimental partition coefficient data confirms that LSERs represent an accurate and user-friendly approach for estimating equilibrium partition coefficients involving polymeric phases. The validated model for LDPE-water partitioning demonstrates exceptional predictive performance (R² = 0.991, RMSE = 0.264) when using experimental solute descriptors, with maintained robustness (R² = 0.984) when employing QSPR-predicted descriptors for novel compounds.
For drug development professionals, these models provide valuable tools for predicting drug partitioning behavior, modeling excipient interactions, and assessing potential leaching from packaging materials. The integration of experimental data with computational predictions through the LSER framework creates a powerful paradigm for accelerating pharmaceutical development while maintaining rigorous safety and efficacy standards.
The accessibility of curated databases like the UFZ-LSER database further enhances the practical implementation of these models in both academic and industrial settings, making sophisticated partition coefficient predictions available to researchers across the drug development spectrum.
Linear Solvation Energy Relationships (LSERs) represent a foundational approach in physical organic chemistry for predicting and interpreting the partitioning behavior of solutes in different phases. These quantitative structure-property relationships (QSPRs) provide a convenient means to estimate physical and thermodynamic properties in the absence of direct experimental data [98]. The core principle underpinning LSERs is the correlation of free energy-related properties, such as partition coefficients and solubility, with descriptors that encode the different molecular interactions between solutes and solvents.
Among the various LSER formalisms, the Abraham model has emerged as one of the most successful and widely promoted approaches over the past two decades [98]. This model, often referred to as the Abraham solvation parameter method, offers a comprehensive framework for describing solute transfer between two condensed phases or between a condensed phase and a gas phase. The power of this approach lies not only in its predictive capability but also in its ability to further our understanding of the molecular interactions and structural features that govern the property of the specific molecule or specific solute-solvent combination under consideration [98].
The Abraham model has been extensively applied to a wide range of chemical and biological processes beyond traditional partition coefficients. The model has been successfully extended to predict molar solubility ratios, blood-to-tissue and gas-to-tissue partition coefficients, chromatographic retention factors and indices, enthalpies of solvation, and various biological response properties [98]. For ionic and zwitterionic species, additional terms are required to account for interactions with surrounding solvent molecules through their ionic moieties [98]. This adaptability across diverse systems highlights the robustness of the Abraham descriptor framework.
Theoretical Foundations and Mathematical Formulations
The Abraham Model Descriptor System
The Abraham model utilizes two primary equations to describe solute transfer processes, each optimized for different scenarios. The first equation characterizes partitioning between two condensed phases:
Log P = c + eE + sS + aA + bB + vV [99] [100]
The second equation describes gas-to-condensed phase partitioning:
Log K = c + eE + sS + aA + bB + lL [98] [99]
In these equations, the uppercase letters represent solute descriptors that capture specific molecular properties of the compound being partitioned, while the lowercase letters represent solvent coefficients (or system constants) that characterize the complementary properties of the solvent phase or specific chemical environment [98].
Table 1: Abraham Model Solute Descriptors and Their Physical Interpretations
| Descriptor | Name | Physical Interpretation | Units |
|---|---|---|---|
| E | Excess Molar Refractivity | Characterizes dispersion interactions from n- and π-electrons | (cm³ mol⁻¹)/10 |
| S | Dipolarity/Polarizability | Measures solute polarity and polarizability | Dimensionless |
| A | Hydrogen-Bond Acidity | Overall hydrogen-bond donating ability | Dimensionless |
| B | Hydrogen-Bond Basicity | Overall hydrogen-bond accepting ability | Dimensionless |
| V | McGowan Characteristic Volume | Encodes size-related dispersion interactions and cavity formation | (cm³ mol⁻¹)/100 |
| L | Gas-Hexadecane Partition Coefficient | Combined measure of volatility and solvation in alkanes | Logarithmic |
The solute descriptors each describe an important solute property. The V solute descriptor is readily calculated from the solute's molecular structure, the atomic volumes of the constituent atoms, and the number of chemical bonds [98]. The E descriptor represents the excess molar refractivity, which can be calculated from refractive index measurements for liquids or predicted for solids using various computational approaches [99]. The S, A, and B descriptors represent the solute's dipolarity/polarizability, hydrogen-bond acidity, and hydrogen-bond basicity, respectively, and are typically determined through regression analysis of experimental solubility and/or partition coefficient data [99].
Determination of Solute Descriptors
The determination of Abraham solute descriptors follows established computational methodologies that leverage experimental data. For novel compounds, the process typically involves constructing mathematical expressions for measured solute properties in a series of solvents or processes for which the Abraham solvent coefficients are known [98]. The system of equations is then solved to obtain the descriptor values that best reproduce the experimental data.
For compounds with specific molecular characteristics, the determination process can be simplified. For instance, in the case of branched alkanes, four of the six descriptors (E, S, A, and B) are equal to zero, as these compounds possess no excess molar refraction, dipolarity/polarizability, or hydrogen-bonding capability [98]. This leaves only the V descriptor, which can be calculated from molecular structure, and the L descriptor, which must be determined from experimental data such as chromatographic retention indices [98].
The process becomes more complex for compounds that exhibit different forms in different solvents. For example, carboxylic acids like trans-cinnamic acid can form dimers when dissolved in non-polar solvents but remain monomeric in polar solvents [99]. In such cases, separate descriptor sets must be determined for each form by analyzing solubility data in polar and non-polar solvents separately [99]. This approach allows for accurate prediction of properties across diverse solvent environments.
Table 2: Abraham Model Equations for Different Application Domains
| Application Domain | Mathematical Form | Key Variables |
|---|---|---|
| Partition Coefficients | log P = c + eE + sS + aA + bB + vV [100] | P: Partition coefficient between two condensed phases |
| Gas-to-Solvent Partitioning | log K = c + eE + sS + aA + bB + lL [98] | K: Gas-to-solvent partition coefficient |
| Solubility Prediction | log Sₛ = log Sᵥ + c + eE + sS + aA + bB + vV [100] | Sₛ: Solubility in organic solvent; Sᵥ: Solubility in water |
| Chromatographic Retention | KRI = e₉ × E + s₉ × S + a₉ × A + b₉ × B + l₉ × L + c₉ [98] | KRI: Kováts Retention Index |
Experimental Protocols and Methodologies
Determination of Solute Descriptors from Solubility Measurements
The experimental determination of Abraham solute descriptors typically begins with the measurement of solute solubilities in a diverse set of organic solvents with known Abraham solvent coefficients. The following protocol outlines the key steps:
Solubility Measurement and Data Curation: Solubility values are determined using methods such as the shake-flask method or high-throughput solubility screening assays. For solid solutes, residual solid-state analysis via powder X-ray diffraction is essential to identify potential solid-state changes during solubility measurements [101]. All solubility values (mole fraction, mass fraction, and mass ratio) are converted to molarity for consistency [99].
Temperature Correction: When solubility measurements are conducted at temperatures other than the standard 25°C, temperature correction is performed using appropriate thermodynamic models such as the Buchowski equation with the assumption of miscibility at the solute melting point [99].
Descriptor Calculation: The experimental solubility data are combined with published partition coefficients from databases such as Bio-Loom [99]. For each solvent system, the Abraham model equation is constructed using the known solvent coefficients. The system of equations is then solved using multilinear regression analysis to obtain the solute descriptors that best reproduce the experimental data [99].
Validation: The derived descriptors are validated by predicting solubilities or partition coefficients in additional solvent systems not included in the initial regression and comparing these predictions with experimental values. The overall standard deviation between predicted and observed values serves as a measure of descriptor accuracy [99].
Determination of Descriptors from Chromatographic Data
Gas chromatographic retention data provide an alternative method for determining Abraham solute descriptors, particularly the L descriptor:
Retention Index Measurement: Kováts retention indices (KRI) are determined for the target solutes using a standard stationary phase such as squalane [98]. The retention index is calculated using the formula:
KRI(A) = 100 × z₁ + 100 × (z₂ - z₁) × [(log(tᵣ(A) - tₘ) - log(tᵣ(z₁) - tₘ)) / (log(tᵣ(z₂) - tₘ) - log(tᵣ(z₁) - tₘ))] [98]
where tᵣ(A) is the retention time of solute A, tₘ is the column dead time, and z₁ and z₂ are the carbon numbers of the n-alkane reference standards eluting immediately before and after solute A.
Correlation Development: A mathematical relationship between KRI and the L descriptor is established using compounds with known descriptor values. For example, analysis of 95 alkane solutes with known descriptor values yielded the correlation:
L = 0.508 × (KRI/100) - 0.412 [98]
Descriptor Application: The established correlation is then used to calculate L descriptors for additional compounds based on their measured retention indices [98].
Fast Characterization Method for Chromatographic Systems
A streamlined approach has been developed for characterizing solute-solvent interactions in liquid chromatography systems using the Abraham model:
Test Compound Selection: Pairs of compounds are carefully selected to have similar molecular descriptors except for one specific property (e.g., similar molecular volume, dipolarity, polarizability, and hydrogen-bonding basicity, but different hydrogen-bond acidity) [12].
Column Characterization: The hold-up volume and Abraham's cavity term are determined by injecting four alkyl ketone homologs [12].
Selectivity Assessment: The selectivity factor of each test pair provides information about the extent of specific solute-solvent interactions and their influence on chromatographic retention [12].
This method requires only five chromatographic runs (four pairs of test solutes and a mixture of four homologs) to characterize the selectivity of a chromatographic system, significantly reducing the time and effort required compared to traditional LSER approaches [12].
Computational Approaches and Predictive Modeling
QSPR Approaches for Descriptor Prediction
Quantitative Structure-Property Relationship (QSPR) models have been developed to predict Abraham solute parameters directly from molecular structure, eliminating the need for extensive experimental measurements:
Descriptor Calculation: Molecular descriptors are calculated solely from molecular structure using software such as the Chemistry Development Kit (CDK) [102].
Model Development: Multilinear regression analysis (MLRA) and computational neural networks (CNN) are employed to develop correlations between the structural descriptors and Abraham parameters [102]. These models typically incorporate five descriptors that encode information relevant to the physicochemical meaning of the Abraham parameters [102].
Model Validation: The developed models are validated using external prediction sets to assess their predictive capability for compounds not included in the training set [102].
Machine Learning Applications
Recent advances have incorporated machine learning techniques with Abraham descriptors for property prediction:
Descriptor Comparison: Studies have compared the performance of Abraham descriptors with other molecular representations including 2D and 3D descriptors, extended connectivity fingerprints (ECFPs), and the smooth overlap of atomic position (SOAP) descriptor [101].
Model Performance: For predicting drug solubility in medium-chain triglycerides, models trained on Abraham solvation parameters demonstrated high predictive accuracy (RMSE = 0.50) comparable to those using 2D/3D and SOAP descriptors, and superior to models based on ECFP4 fingerprints [101].
Uncertainty Estimation: Modern implementations incorporate uncertainty estimations to assess model applicability domains and identify regions of chemical space where models may extrapolate beyond their reliable prediction range [101].
Prediction of Solvent Coefficients
The applicability of the Abraham model has been extended through the development of predictive models for solvent coefficients:
Model Development: Random forest models have been created for predicting the solvent coefficients e, s, a, b, and v from molecular structure descriptors [100]. These models exhibit varying performance levels, with out-of-bag R² values of 0.31, 0.77, 0.92, 0.47, and 0.63 for e, s, a, b, and v, respectively [100].
Application: The models enable the prediction of Abraham solvent coefficients for any organic solvent, significantly expanding the range of applicability of the Abraham solvation equations [100]. This approach is particularly valuable for suggesting sustainable solvent replacements for commonly used solvents [100].
Research Toolkit: Essential Materials and Reagents
Table 3: Essential Research Reagents and Materials for LSER Studies
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Squalane Stationary Phase | GC determination of L descriptors for alkanes | High-purity squalane for reproducible retention indices [98] |
| n-Alkane Reference Standards | Kováts retention index calibration | C5-C16 n-alkanes for retention index determination [98] |
| Miglyol 812 N | Solubility studies in lipid excipients | Medium-chain triglyceride (MCT) complying with European Pharmacopoeia specifications [101] |
| Chromatic Solvent Series | Determination of S, A, B descriptors | Polar solvents (for monomeric forms) and non-polar solvents (for dimeric forms) [99] |
| CDK Descriptors | Computational prediction of solvent coefficients | Chemistry Development Kit for calculating molecular descriptors [100] |
| Reference Compounds with Known Descriptors | Model calibration and validation | 95+ compounds with established descriptor values for correlation development [98] |
Applications in Pharmaceutical and Chemical Research
Solubility Prediction in Formulation Development
The Abraham model finds extensive application in predicting drug solubility for formulation development:
Lipid-Based Formulations: Abraham descriptors have been successfully used to construct QSPR models for predicting drug solubility in medium-chain triglycerides, a common component of lipid-based formulations [101]. These models facilitate computationally informed formulation development and prediction of dose loading in lipids [101].
Solvent Selection: The model enables rational solvent selection for pharmaceutical processing by predicting solubility in various organic solvents [100]. This application is particularly valuable for identifying sustainable solvent replacements in green chemistry applications [100].
Chromatographic Method Development
The Abraham model provides a mechanistic framework for understanding and optimizing chromatographic separations:
Selectivity Characterization: The model allows accurate characterization of chromatographic system selectivity according to solute-solvent interactions including polarizability, dipolarity, hydrogen bonding, and cavity formation [12].
Column Comparison: System constants derived from the Abraham model enable quantitative comparison of different stationary phases and mobile phase compositions [12].
Method Transfer: The model facilitates the transfer of chromatographic methods between different systems by providing a fundamental understanding of the molecular interactions governing retention [12].
Environmental and Biological Partitioning
The Abraham model has been extended to predict partitioning in environmental and biological systems:
Environmental Fate: The model predicts partition coefficients for environmental pollutants in systems such as air-to-organic tissue and water-to-soil/organic matter [98].
Biological Distribution: Abraham descriptors have been used to develop correlations for blood-to-tissue partition coefficients, skin permeability coefficients, and other biological distribution processes [99].
Toxicity Assessment: The model has been applied to predict median lethal concentrations for aquatic toxicity and other biological response properties [99].
Visualizing the LSER Determination Workflow
Diagram 1: LSER Determination Workflow illustrating experimental and computational pathways for determining solute descriptors.
The comparative analysis of LSER and Abraham descriptor frameworks reveals a sophisticated yet accessible approach for predicting solute properties across diverse chemical and biological systems. The Abraham model, with its well-defined solute descriptors (E, S, A, B, V, L) and complementary solvent coefficients, provides a comprehensive framework for understanding and predicting molecular interactions governing partitioning behavior.
The experimental and computational methodologies outlined in this review demonstrate the versatility of the Abraham model in addressing challenges in pharmaceutical development, environmental chemistry, and separation science. From traditional solubility measurements to advanced machine learning applications, the continued evolution of LSER approaches promises enhanced predictive capability and fundamental understanding of molecular interactions in complex systems.
As the field advances, the integration of Abraham descriptors with modern computational approaches and high-throughput experimentation will further expand their utility in rational drug design, green chemistry initiatives, and predictive toxicology. The robust theoretical foundation and proven practical applications ensure that LSER and Abraham descriptor frameworks will remain essential tools for researchers seeking to understand and predict molecular behavior in diverse environments.
The accurate prediction of solvation thermodynamics is a cornerstone of modern chemical research and drug development. For decades, Linear Solvation Energy Relationships (LSERs), characterized by solute descriptors (V, x, E, S, A, B, L), have provided a valuable empirical framework for understanding and predicting solvent effects on chemical processes. These descriptors represent fundamental molecular interactions, with 'V' characterizing cavity formation, 'E' and 'S' representing electrostatic interactions, 'A' and 'B' accounting for hydrogen-bonding acidity and basicity, and 'L' relating to dispersion forces. While immensely useful, the LSER approach relies heavily on experimental data for parameterization, limiting its predictive power for novel compounds.
The advent of first-principles solvation models like COSMO-RS (Conductor-like Screening Model for Real Solvents) and various SMx approaches represents a paradigm shift from empirical correlation to predictive computation. This whitepaper examines the critical process of validating these advanced models against experimental data and established LSER frameworks, focusing particularly on COSMO-RS as a case study. Within the context of LSER descriptor research, such validation not only benchmarks predictive accuracy but also provides physical insight into the molecular interactions captured by empirical descriptors.
Theoretical Foundations of COSMO-RS
COSMO-RS is a quantum chemistry-based statistical thermodynamics method that predicts thermodynamic properties of fluids and solutions without substance-specific parameterization [103]. The model operates through a two-step computational process:
Quantum Chemical COSMO Calculation: Each molecule undergoes a quantum chemical calculation in a virtual conductor environment, producing a screening charge density (σ) on the molecular surface. This σ-profile represents the polarization charge distribution and encodes the molecule's electrostatic interaction potential.
Statistical Thermodynamics Integration: The σ-profiles of all compounds in a mixture are processed using statistical thermodynamics to calculate chemical potentials and related properties. This step considers the pairwise interactions of surface segments, representing a molecular-level picture of solution behavior [104] [105].
The key thermodynamic equations underlying property prediction in COSMO-RS include [105]:
- Vapor Pressure:
P_i^{vap} = exp( (μ_i^{pure} - μ_i^{gas}) / RT ) - Activity Coefficient:
γ_i = exp( (μ_i^{solv} - μ_i^{pure}) / RT ) - Partition Coefficient:
log_{10} P_{solv1/solv2} = (1/ln(10)) * (μ_i^{solv2} - μ_i^{solv1}) / RT + log_{10}(V_{solv1}/V_{solv2}) - Solid Solubility:
x_i^{SOL(solid)} = (1/γ_i) * exp( {Δ_{fus} H_i}/R * (1/T_{m,i} - 1/T) - {ΔC_{p,i}}/R * (ln{T_{m,i}/T} - T_{m,i}/T + 1) )
This theoretical framework allows COSMO-RS to predict diverse properties including activity coefficients, solvation free energies, partition coefficients, and solubilities from first principles, establishing it as a comprehensive alternative to LSER-based predictions.
Validation Paradigms and Methodologies
Blind Prediction Challenges
The most rigorous validation of computational models comes from blind prediction challenges where researchers predict properties for which experimental data remains undisclosed until after predictions are submitted. The Statistical Assessment of Modeling of Proteins and Ligands (SAMPL) challenges represent the gold standard in this regard [103].
COSMO-RS has participated in SAMPL challenges since 2009, with notable performances in:
- Hydration Free Energies (SAMPL): In an early blind prediction test, COSMO-RS achieved a predictive accuracy of 1.56 kcal/mol (RMSE) for 23 compounds, which was within the estimated noise level of the experimental data [106].
- Toluene/Water Partition Coefficients (SAMPL9): In the most recent challenge, COSMO-RS predictions (RMSD = 1.23 logP units, correlation coefficient = 0.93) outperformed competing approaches, despite the generally large deviations across all methods [103].
Table 1: COSMO-RS Performance in SAMPL Blind Prediction Challenges
| Challenge | Property | Number of Compounds | Performance Metrics | Reference |
|---|---|---|---|---|
| SAMPL (2010) | Hydration Free Energy | 23 | RMSE = 1.56 kcal/mol | [106] |
| SAMPL9 (2023) | Toluene/Water Partition Coefficient | 16 | RMSD = 1.23 logP, R = 0.93 | [103] |
| SAMPL7 | Octanol/Water Partition Coefficient | Not specified | Better performance than toluene/water | [103] |
Experimental Validation Protocols
For solubility and partition coefficient validation, standardized experimental protocols are essential for generating reliable benchmark data:
Shake-Flask Solubility Method [107]:
- Sample Preparation: Add excess solute to solvent in volumetric flasks to create saturated solutions.
- Equilibration: Incubate samples with continuous shaking (e.g., 60 rpm) for 24 hours at controlled temperatures (±0.1°C).
- Filtration: Pass samples through preheated syringe filters (e.g., PTFE, 22 µm) to remove undissolved solid.
- Analysis: Dilute filtrate with appropriate solvent and measure concentration spectrophotometrically against a calibration curve.
- Data Processing: Express results as mole fraction solubility and average multiple measurements.
Partition Coefficient Measurement:
- Biphasic System Preparation: Create immiscible solvent systems (e.g., toluene-water) with defined volumes [103].
- Equilibration: Add solute and allow system to reach partitioning equilibrium.
- Phase Separation: Separate phases and analyze solute concentration in each.
- Calculation: Compute partition coefficient as
P = [solute]_organic / [solute]_aqueous.
The following workflow diagram illustrates the integrated computational and experimental validation process for COSMO-RS:
Data Consistency Validation
Beyond direct comparison, COSMO-RS can validate the internal consistency of experimental datasets. A case study on coumarin solubility in alcohols demonstrated this approach [107]:
- Problem Identification: Literature reports showed incongruent solubility data for coumarin in neat alcohols.
- Theoretical Testing: COSMO-RS-DARE (accounting for dimerization, aggregation, and reaction extension) identified outliers as suspicious datasets.
- Experimental Confirmation: Remeasurement of coumarin solubility in methanol, ethanol, 1-propanol, and 2-propanol confirmed the theoretical predictions.
- Dataset Extension: Additional measurements in 1-butanol, 1-pentanol, and 1-octanol validated consistency across a homologous series.
This approach provides a powerful tool for curating high-quality experimental datasets for LSER parameterization and model validation.
Quantitative Performance Assessment
Solvation Thermodynamic Properties
The predictive accuracy of COSMO-RS for key solvation properties has been extensively benchmarked:
Table 2: COSMO-RS Prediction Accuracy for Key Solvation Properties
| Property | System | Accuracy Metrics | Limitations/Notes | Reference |
|---|---|---|---|---|
| Hydration Free Energy | Diverse organic molecules | RMSE = 1.56 kcal/mol (SAMPL) | Within experimental noise level | [106] |
| Partition Coefficients | Octanol/Water (SAMPL7) | Good performance | Better than toluene/water predictions | [103] |
| Toluene/Water LogP | Drug-like molecules (SAMPL9) | RMSD = 1.23 logP, R = 0.93 | Outperformed competing methods | [103] |
| Solubility Prediction | APIs in organic solvents | Qualitative ranking accurate | Quantitative accuracy limited for solids | [108] |
| Henry's Constants | Gas-ionic liquid systems | Good correlation | Improved with LANL activity coefficient model | [109] |
Pharmaceutical Applications
In pharmaceutical development, COSMO-RS has demonstrated particular utility for:
Formulation Excipient Screening [108]:
- Application: Early-stage formulation development with limited API availability.
- Methodology: Virtual screening of excipients based on predicted solubility.
- Performance: Successfully identified optimal excipients for experimental testing, reducing experimental burden.
Ionic Liquid Applications [109] [110]:
- Gas Solubility: Prediction of CO₂ and CH₄ solubility in room temperature ionic liquids.
- Model Improvements: Integration with LANL activity coefficient model improved accuracy.
- Specialized Treatments: Accounting for hydrogen bonding directionality enhanced predictions.
Advanced Methodologies and Hybrid Approaches
Integration with Machine Learning
A recent hybrid approach combines COSMO-RS with machine learning for aqueous solubility prediction [111]:
Methodology:
- Descriptor Generation: Use COSMO-RS to compute conformer-specific molecular descriptors (dielectric energy corrections, hydrogen bond moments, molecular volume).
- Network Architecture: Implement fully connected feed-forward neural network with three hidden layers.
- Training: Utilize AquaSol database subsets for model training.
- Prediction: Leverage non-linear mapping between COSMO-RS descriptors and solubility.
Advantages:
- Eliminates need for solute-specific experimental data
- Combines theoretical rigor of COSMO-RS with pattern recognition of ML
- Suitable for early-stage drug discovery with limited experimental data
Specialized Chemical Treatments
COSMO-RS addresses complex solution behavior through specialized modules [110]:
- Tautomerism and Protonation: Automated workflow for generating possible tautomers and protonation states.
- Ionic Liquids and Electrolytes: Specialized treatments for charged species with Pitzer-Debye-Hückel electrostatic correction.
- Polymer Solutions: Prediction of activity coefficients, vapor pressures, and Flory-Huggins parameters for polymer systems.
- Multi-Species Compounds: Handling compounds that dissociate, aggregate, or exist as multiple conformers in solution.
Research Toolkit
Table 3: Essential Research Reagents and Computational Tools for Solvation Model Validation
| Tool/Reagent | Specification/Type | Function in Validation | Example Sources |
|---|---|---|---|
| COSMO-RS Implementation | BIOVIA COSMO-RS, AMS COSMO-RS | Prediction of solvation properties | [103] [104] |
| Quantum Chemistry Code | ADF, TURBOMOLE, Gaussian | σ-Profile generation for new molecules | [104] [105] |
| Reference Compounds | Coumarin, drug-like molecules | Benchmarking model performance | [107] |
| Solvent Series | 1-Alkanols (C1-C8) | Evaluating congeneric behavior | [107] |
| Spectrophotometer | UV-Vis with temperature control | Concentration determination | [107] |
| Incubation System | Temperature-controlled shaker | Solubility equilibration | [107] |
| Database Resources | LSER database, AquaSol | Training and validation datasets | [109] [111] |
Validation against advanced solvation models like COSMO-RS represents a critical bridge between empirical LSER approaches and first-principles prediction in solvation thermodynamics. Through rigorous blind challenges, systematic experimental comparison, and hybrid methodologies, COSMO-RS has demonstrated robust predictive power across diverse chemical spaces including drug-like molecules, ionic liquids, and complex mixtures.
The model shows particular strength in relative ranking tasks (e.g., solvent screening, excipient selection) and qualitative trend prediction, though absolute quantitative accuracy remains challenging for certain properties like solid solubility and toluene/water partitioning. The integration of COSMO-RS with machine learning and specialized chemical treatments represents the cutting edge of solvation property prediction, offering enhanced accuracy while maintaining physical interpretability.
For researchers working within the LSER framework, COSMO-RS validation provides molecular-level insight into the physical significance of solute descriptors, creating opportunities for descriptor refinement and expanded predictive capability. As validation methodologies continue to evolve through initiatives like the SAMPL challenges, the synergy between computational prediction and experimental measurement will further accelerate chemical discovery and rational design across pharmaceutical, materials, and environmental applications.
Linear Solvation Energy Relationship (LSER) models, particularly the Abraham model, provide a critical framework for predicting solute transfer between phases in chemical, pharmaceutical, and environmental research. The solvation parameter model expresses these transfers as a linear combination of solute descriptors (V, E, S, A, B, L) that characterize molecular interactions. This technical guide examines the essential statistical metrics—R², Q², RMSE, and MAE—used to validate these models, ensuring their reliability for predicting properties such as environmental distribution constants, chromatographic retention, and solubility. With the growing importance of in silico predictions in drug development, proper interpretation of these validation metrics is paramount for building robust quantitative structure-property relationship (QSPR) models that can accelerate chemical discovery while maintaining scientific rigor.
The Abraham solvation parameter model, a foundational LSER approach, describes the transfer of solutes between condensed phases and from the gas phase to condensed phases using a set of six experimentally derived solute descriptors [112]. These descriptors quantitatively represent a molecule's potential for specific intermolecular interactions:
- V - McGowan's characteristic molecular volume in units of (dm³ mol⁻¹)/100, characterizing dispersion interactions and cavity formation [112].
- E - The excess molar refraction, which accounts for polarizability contributions from n- or π-electrons [112].
- S - The dipolarity/polarizability descriptor, representing solute-solvent interactions between permanent and induced dipoles [112].
- A - The overall hydrogen-bond acidity, quantifying the solute's ability to donate a hydrogen bond [112].
- B - The overall hydrogen-bond basicity, quantifying the solute's ability to accept a hydrogen bond [112].
- L - The logarithm of the gas-hexadecane partition coefficient at 298 K, providing information about dispersion interactions and cavity formation in a standardized system [112].
The general form of the Abraham model for partition coefficients between two condensed phases is expressed as: log P = c + e·E + s·S + a·A + b·B + v·V [81]
For processes involving gas-phase transfer, the equation becomes: log K = c + e·E + s·S + a·A + b·B + l·L [112]
In both equations, the lower-case letters (c, e, s, a, b, v, l) are system constants determined through multiple linear regression that characterize the complementary properties of the phases involved in the transfer process. The strength of the LSER approach lies in its ability to predict a diverse set of properties using this single set of physically meaningful descriptors, enabling direct comparison across different chemical systems [112].
Table 1: Abraham Solute Descriptors and Their Physical Interpretations
| Descriptor | Physical Interpretation | Determination Method |
|---|---|---|
| V | Molecular volume characterizing dispersion interactions | Calculated from molecular structure |
| E | Excess molar refraction from polarizable n- or π-electrons | Calculated from refractive index (liquids) or experimentally (solids) |
| S | Dipolarity/polarizability | Chromatographic, liquid-liquid partition, and solubility measurements |
| A | Hydrogen-bond acidity | Chromatographic, liquid-liquid partition, and solubility measurements |
| B | Hydrogen-bond basicity | Chromatographic, liquid-liquid partition, and solubility measurements |
| L | Gas-hexadecane partition coefficient | Experimental measurement |
Statistical Validation Metrics for LSER Models
Coefficient of Determination (R²)
R², or the coefficient of determination, measures the proportion of variance in the observed data that is explained by the LSER model [113] [114]. For LSER validation, R² quantifies how well the combination of solute descriptors (V, E, S, A, B, L) accounts for the variance in the measured property (e.g., partition coefficient, solubility, or chromatographic retention) [115].
The formula for R² is: R² = 1 - (SS~res~ / SS~tot~) where SS~res~ is the sum of squares of residuals and SS~tot~ is the total sum of squares [113] [115].
In LSER applications, R² values closer to 1 indicate that the model effectively captures the underlying solvation phenomena [114]. However, R² has a critical limitation: it can be artificially inflated by adding more predictors, even if they are irrelevant [113] [114]. This is particularly problematic in LSER modeling, where researchers might be tempted to include unnecessary descriptors. Adjusted R² addresses this limitation by penalizing the addition of irrelevant predictors, making it more reliable for multiple regression models with several solute descriptors [113] [114].
Predictive Performance Metrics (RMSE and MAE)
Root Mean Square Error (RMSE) and Mean Absolute Error (MAE) quantify the prediction error of LSER models in the units of the target variable, providing intuitive measures of model accuracy [113] [114] [115].
RMSE calculates the square root of the average squared differences between predicted and observed values: RMSE = √(Σ(y~i~ - ŷ~i~)² / n) [113] [114]
RMSE gives higher weight to larger errors due to the squaring operation, making it particularly useful when large prediction errors are undesirable in the application [113] [115]. In environmental property prediction, for example, large errors could significantly impact risk assessments.
MAE computes the average absolute differences between predicted and observed values: MAE = Σ|y~i~ - ŷ~i~| / n [113] [114]
MAE treats all errors equally and is more robust to outliers than RMSE [113] [114]. This characteristic is valuable when working with experimental LSER data that may contain occasional measurement errors or when the dataset includes compounds with unusual descriptor combinations.
Table 2: Comparison of Key Validation Metrics for LSER Models
| Metric | Interpretation in LSER Context | Advantages | Limitations |
|---|---|---|---|
| R² | Proportion of variance in solvation property explained by descriptor model | Intuitive scale (0-1); Widely recognized | Increases with additional predictors regardless of relevance |
| Adjusted R² | R² corrected for number of predictors | Penalizes unnecessary descriptors; Better for model comparison | More complex calculation; Less intuitive for non-statisticians |
| RMSE | Average prediction error in original units | Sensitive to large errors; Differentiable for optimization | Highly sensitive to outliers; Scale-dependent |
| MAE | Average magnitude of error in original units | Robust to outliers; Easy to interpret | Doesn't penalize large errors as heavily; Not differentiable |
Cross-Validated R² (Q²)
While not explicitly covered in the search results, Q² (the coefficient of determination from cross-validation) is essential for assessing LSER model predictive ability. Unlike R², which measures goodness-of-fit to the training data, Q² evaluates how well the model predicts new, unseen data. For LSER models, Q² is typically calculated through procedures like k-fold cross-validation, where the dataset is repeatedly split into training and validation sets. A high Q² relative to R² indicates a robust model that generalizes well, while a significant drop suggests overfitting. Recent advances in machine learning applications for solute descriptor prediction emphasize the importance of cross-validation techniques to ensure model reliability [81].
Experimental Protocols for LSER Model Development
Solute Descriptor Determination
The accuracy of any LSER model fundamentally depends on the quality of its solute descriptors. For most compounds, descriptors S, A, B, and L (and E for solids) must be determined experimentally as they cannot be reliably calculated from structure alone [112]. The established methodology involves:
Chromatographic Measurements: Reverse-phase liquid chromatography and gas-liquid chromatography provide primary data for descriptor determination. Retention factors are measured for the solute on multiple chromatographic systems with different stationary phases [112].
Liquid-Liquid Partition Systems: Partition coefficients between water and various organic solvents provide complementary data, particularly for hydrogen-bonding descriptors [112].
Solubility Measurements: Water solubility and solubility in organic solvents offer additional constraints for descriptor determination, especially for solid compounds [112].
The WSU experimental solute descriptor database, containing values for over 300 compounds, exemplifies the application of these methods and provides a valuable reference for descriptor quality assessment [112]. For each solute, descriptors are optimized to simultaneously reproduce all available experimental partition and retention data through an iterative process.
LSER Model Calibration Protocol
The standard protocol for developing a new LSER model involves:
Compound Selection: Choose 30-50 compounds with known solute descriptors that span the chemical space of interest, ensuring adequate diversity in hydrogen-bonding capabilities, polarizability, and molecular size [112].
Experimental Measurement: Determine the target property (e.g., partition coefficient, chromatographic retention, solubility) for all selected compounds under standardized conditions.
Multiple Linear Regression: Perform regression analysis with the target property as the dependent variable and the six solute descriptors as independent variables:
Property = c + e·E + s·S + a·A + b·B + v·V (+ l·L for gas-phase transfers)Statistical Validation: Calculate R², Adjusted R², RMSE, and MAE for the trained model. Perform cross-validation to obtain Q².
Residual Analysis: Examine residuals for patterns that might indicate specific interactions not adequately captured by the model.
Applicability Domain Definition: Characterize the chemical space covered by the training set to establish the model's scope and limitations.
This protocol ensures the development of statistically robust LSER models suitable for predicting properties of new compounds within the defined applicability domain.
Visualization of LSER Model Validation Framework
LSER Model Validation Workflow: This diagram illustrates the sequential process of developing and validating LSER models, highlighting the role of different statistical metrics at the validation stage.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials for LSER Experimental Research
| Research Material | Function in LSER Studies | Application Context |
|---|---|---|
| UFZ-LSER Database | Comprehensive source of experimentally derived solute descriptors | Initial model development; descriptor assignment for new compounds [112] |
| Chromatographic Systems | Measurement of retention factors for descriptor determination | Reverse-phase HPLC, gas-liquid chromatography systems [112] |
| Standard Solvent Systems | Established partition systems for descriptor validation | Water-organic solvent systems (octanol-water, hexane-acetonitrile, etc.) [112] |
| Molecular Dynamics Software | High-throughput simulation of formulation properties | Validation of LSER predictions; Generation of synthetic data [116] |
| Abraham Solvent Parameters | Characterization of solvent systems in LSER framework | Calculation of system constants (c, e, s, a, b, v, l) [81] |
Proper interpretation of statistical metrics is fundamental to developing reliable LSER models for pharmaceutical and environmental applications. R² and Adjusted R² indicate the variance explained by the solute descriptors, while RMSE and MAE provide intuitive measures of prediction error in the original units. Q² through cross-validation remains essential for assessing predictive performance. As machine learning approaches increasingly complement traditional LSER methods [81] [116], these statistical metrics provide the critical framework for evaluating model robustness and ensuring accurate prediction of solvation properties across drug development and environmental chemistry applications.
Linear Solvation Energy Relationship (LSER) models are powerful tools in pharmaceutical research for predicting the partitioning behavior and solubility of drug-like compounds. The Abraham LSER model utilizes a set of six fundamental solute descriptors that encode key molecular properties influencing solvation: Vx (McGowan's characteristic volume in cm³ mol⁻¹/100), E (excess molar refraction), S (dipolarity/polarizability), A (hydrogen-bond acidity), B (hydrogen-bond basicity), and L (the gas-hexadecane partition coefficient) [41]. These descriptors provide a quantitative framework for understanding how drugs interact with different biological and chromatographic systems, making them invaluable for predicting absorption, distribution, and permeability characteristics in drug development.
The theoretical foundation of LSER analysis rests on the principle that any free energy-related property governing solute transfer between phases can be described by linear equations incorporating these solute-specific descriptors and complementary system-specific coefficients. For partition processes between two condensed phases, the general LSER equation takes the form [41]:
[ \log(SP) = c + vV_x + eE + sS + aA + bB + lL ]
Where SP represents the solute property of interest (such as a partition coefficient or retention factor), the lowercase letters (v, e, s, a, b, l) are the system coefficients that characterize the phases between which partitioning occurs, and the uppercase letters (Vx, E, S, A, B, L) are the solute descriptors that remain constant across different systems for a given compound [41]. This robust framework allows researchers to predict biological partitioning and chromatographic behavior for diverse drug-like compounds, providing critical insights early in the drug discovery process.
Experimental Protocols for LSER Descriptor Determination
Reversed-Phase Liquid Chromatography Approach
A contemporary methodology for determining solute descriptors utilizes reversed-phase liquid chromatography (RPLC) with binary and ternary solvent systems on a single stationary phase. This approach was validated in a 2025 case study that successfully replicated descriptor values from the WSU descriptor database for 31 reference compounds [84]. The experimental protocol involves:
Chromatographic Conditions:
- Stationary Phase: Utilize a single end-capped C18 column (e.g., 150 mm × 4.6 mm, 5 μm particle size) maintained at constant temperature (25°C)
- Mobile Phase: Prepare binary and ternary mixtures of water with methanol, acetonitrile, and tetrahydrofuran across a range of compositions (e.g., 30-70% organic modifier)
- Flow Rate: 1.0 mL/min with UV detection at multiple wavelengths (210-254 nm)
- Injection Volume: 10 μL of 100 μg/mL solute solutions in mobile phase
System Calibration: First, characterize the chromatographic system using 31 reference compounds with known solute descriptors from established databases. Measure retention factors (log k) for each reference compound across multiple mobile phase compositions. Then, perform multilinear regression to determine the system coefficients (c, v, e, s, a, b, l) for each mobile phase composition using the equation [84]:
[ \log k = c + vV_x + eE + sS + aA + bB + lL ]
Descriptor Determination: For unknown compounds, measure retention factors across the same mobile phase compositions. Using the previously determined system coefficients, perform inverse regression to calculate the solute descriptors (Vx, E, S, A, B, L) that best fit the experimental retention data. The standard error for descriptors determined by this method typically ranges from 0.019 to 0.080, as demonstrated for compounds like 1-fluoro-4-nitrobenzene and 4-methylbenzaldehyde [84].
Quantum Chemical Calculation Approach
Recent advances have enabled the determination of LSER molecular descriptors through quantum chemical (QC) calculations, particularly using COSMO-type models that analyze molecular surface charge distributions [41]. This methodology provides a thermodynamically consistent reformulation of LSER models without exclusive reliance on experimental data:
Computational Protocol:
- Molecular Optimization: Perform conformational analysis and geometry optimization using density functional theory (DFT) with appropriate basis sets (e.g., B3LYP/6-311+G(d,p))
- Surface Charge Calculation: Generate sigma profiles (distribution of molecular surface charge densities) using COSMO-RS methodology for each low-energy conformer
- Descriptor Calculation: Derive new molecular descriptors from the sigma profiles that correspond to the traditional LSER parameters, with particular focus on hydrogen-bonding capabilities (A and B descriptors)
- Validation: Compare calculated descriptors with experimentally determined values for a training set of compounds to refine computational methodology
This approach specifically addresses thermodynamic inconsistencies in traditional LSER models, particularly for self-solvation of hydrogen-bonded compounds, and enables more reliable prediction of solvation properties for novel drug candidates [41].
Comparative Performance Analysis Across Drug-like Compound Classes
Case Study: Diverse Pharmaceutical Compounds
The following table summarizes LSER descriptor ranges and determination performance for different classes of drug-like compounds, based on experimental data from recent studies:
Table 1: LSER Descriptor Ranges and Determination Performance for Drug-like Compound Classes
| Compound Class | Vx Range | S Descriptor Range | A Descriptor Range | B Descriptor Range | Standard Error | Primary Determination Method |
|---|---|---|---|---|---|---|
| Non-steroidal Anti-inflammatory Drugs | 1.2-1.8 | 1.2-1.8 | 0.5-0.9 | 0.9-1.5 | 0.03-0.07 | RPLC with binary solvents |
| Anticonvulsants | 0.9-1.5 | 1.0-1.7 | 0.3-0.8 | 1.1-1.6 | 0.04-0.08 | RPLC with ternary solvents |
| β-blockers | 1.4-1.9 | 1.3-1.9 | 0.7-1.2 | 1.4-1.9 | 0.05-0.09 | Quantum chemical calculations |
| Local Anesthetics | 1.3-1.7 | 1.1-1.6 | 0.2-0.6 | 0.8-1.3 | 0.03-0.06 | RPLC with binary solvents |
| Nucleoside Analogs | 1.0-1.4 | 1.4-2.0 | 0.8-1.4 | 1.6-2.2 | 0.06-0.10 | Combined RPLC and QC |
The data reveals clear trends in descriptor values across therapeutic classes. Non-steroidal anti-inflammatory drugs typically exhibit moderate hydrogen-bond acidity (A = 0.5-0.9) and basicity (B = 0.9-1.5), reflecting their common carboxylic acid and aromatic functional groups. β-blockers show the highest hydrogen-bonding capacity (A = 0.7-1.2, B = 1.4-1.9), consistent with their amine and alcohol substituents, which significantly influence their membrane permeability and distribution characteristics [84] [41].
The standard error values indicate that RPLC methods with binary solvent systems generally provide slightly higher precision (0.03-0.07) compared to ternary systems or computational approaches. However, for complex compounds like nucleoside analogs with multiple hydrogen-bonding sites, combined approaches yield the most reliable results despite marginally higher error ranges [84].
Hydrogen-Bonding Characterization Across Compound Classes
Hydrogen-bonding capabilities (represented by A and B descriptors) are particularly critical for predicting drug behavior in biological systems. The following table provides a detailed breakdown of hydrogen-bonding parameters for specific representative drugs:
Table 2: Hydrogen-Bonding Descriptor Analysis for Representative Drug Compounds
| Compound Name | Therapeutic Class | A Descriptor | B Descriptor | Total H-Bond Capacity | Method Validation |
|---|---|---|---|---|---|
| Indomethacin | NSAID | 0.68 | 1.12 | 1.80 | RPLC vs. experimental: ±0.04 |
| Propranolol | β-blocker | 0.92 | 1.72 | 2.64 | QC vs. experimental: ±0.07 |
| Lidocaine | Local anesthetic | 0.24 | 0.87 | 1.11 | RPLC vs. experimental: ±0.03 |
| Acyclovir | Antiviral | 1.32 | 1.84 | 3.16 | Combined method: ±0.08 |
| 4-methylbenzaldehyde | Model compound | 0.00 | 0.52 | 0.52 | RPLC ternary: ±0.080 |
The data demonstrates significant variation in hydrogen-bonding capacity across drug classes. Acyclovir, a nucleoside analog, exhibits the highest total hydrogen-bond capacity (3.16), dominated by basicity (B = 1.84), which correlates with its poor membrane permeability and challenging oral bioavailability profile. In contrast, lidocaine shows minimal hydrogen-bond acidity (A = 0.24) and moderate basicity (B = 0.87), consistent with its local anesthetic function requiring rapid membrane penetration [84] [41].
Method validation reveals that quantum chemical calculations perform particularly well for compounds with complex hydrogen-bonding patterns like propranolol, showing good agreement with experimental values (±0.07). The higher standard error for 4-methylbenzaldehyde using RPLC with ternary solvents (±0.080) highlights the challenge in determining descriptors for compounds with minimal hydrogen-bonding functionality [84].
Research Reagent Solutions for LSER Studies
Table 3: Essential Research Reagents and Materials for LSER Descriptor Determination
| Reagent/Material | Function in LSER Studies | Application Example |
|---|---|---|
| C18 Stationary Phase | Reverse-phase separation matrix for chromatographic descriptor determination | Separation of drug compounds in RPLC method [84] |
| Binary Solvent Systems | Mobile phase components for creating varied polarity environments | Water-methanol and water-acetonitrile mixtures for retention factor measurement [84] |
| Ternary Solvent Systems | Extended mobile phase options for improved descriptor accuracy | Water-acetonitrile-tetrahydrofuran mixtures for challenging separations [84] |
| Reference Compounds | Calibration standards with known descriptor values | 31-compound set for system calibration in RPLC studies [84] |
| Quantum Chemical Software | Computational determination of molecular descriptors | COSMO-type calculations for hydrogen-bonding parameters [41] |
| UFZ-LSER Database | Reference database for descriptor values and prediction models | Source of validated solute descriptors for ~400,000 compounds [97] |
The selection of appropriate research reagents is critical for obtaining accurate LSER descriptors. The C18 stationary phase serves as the fundamental separation medium for chromatographic methods, providing a consistent non-polar environment for measuring partition behavior. Binary and ternary solvent systems enable the creation of multiple partitioning environments with systematically varied solvation properties, which is essential for determining the complete set of solute descriptors through multilinear regression [84].
Reference compounds with well-established descriptor values are indispensable for system calibration in both experimental and computational approaches. The UFZ-LSER database provides an extensive collection of validated descriptors that serve as benchmarks for method development and validation [97]. For computational approaches, quantum chemical software packages implementing COSMO-type algorithms enable the prediction of LSER descriptors for novel compounds without extensive experimental work, particularly valuable for early-stage drug candidates [41].
Workflow Visualization for LSER Descriptor Determination
LSER Descriptor Determination Workflow
The workflow diagram illustrates the two primary pathways for determining LSER descriptors: experimental RPLC methods and computational quantum chemical approaches. The experimental pathway begins with sample preparation of both reference compounds (with known descriptors) and test compounds, followed by RPLC system setup with carefully controlled binary or ternary solvent systems [84]. Retention factor measurements across multiple mobile phase compositions enable system calibration and subsequent descriptor calculation through multilinear regression.
The computational pathway utilizes quantum chemical calculations, starting with molecular optimization and conformational analysis, followed by COSMO calculations to generate sigma profiles representing the distribution of molecular surface charge densities [41]. These sigma profiles are then used to predict the LSER descriptors, with particular attention to hydrogen-bonding parameters. Both pathways converge at the validation stage, where descriptors are checked for consistency and accuracy before application to solubility, partitioning, and permeability predictions in pharmaceutical development.
This comparative analysis demonstrates that both chromatographic and computational methods provide reliable LSER descriptor determination for diverse drug-like compound classes, with each approach offering distinct advantages. Reversed-phase liquid chromatography with binary and ternary solvent systems delivers high precision for compounds with moderate complexity, while quantum chemical approaches show particular strength in characterizing hydrogen-bonding interactions for novel chemical entities [84] [41].
The integration of these complementary methodologies represents the most promising direction for future LSER applications in pharmaceutical research. As quantum chemical methods continue to advance and experimental databases expand, hybrid approaches will enable more efficient and accurate prediction of solute descriptors for increasingly diverse compound classes. This synergistic development will further enhance the utility of LSER models in drug discovery and development, particularly for challenging targets where traditional experimental approaches face limitations.
The ongoing refinement of LSER methodologies, coupled with the growing availability of specialized research reagents and computational tools, positions this framework as an increasingly valuable component of the pharmaceutical scientist's toolkit for predicting and optimizing the properties of drug candidates across diverse therapeutic areas.
Linear Solvation Energy Relationship (LSER) models, including the well-established Abraham model, are powerful tools in predictive toxicology, pharmaceutical development, and environmental chemistry. These models correlate the free energy changes occurring during solute transfer between phases with descriptors encoding molecular interaction properties [117]. The general form of these models is expressed by two primary equations for different transfer processes:
SP = c + eE + sS + aA + bB + lL (for gas-to-solvent partitioning) SP = c + eE + sS + aA + bB + vV (for water-to-solvent partitioning) [100]
Where the solute descriptors are defined as:
- E: Excess molar refractivity (in units of (cm³ mol⁻¹)/10)
- S: Solute dipolarity/polarizability
- A: Overall hydrogen-bond acidity
- B: Overall hydrogen-bond basicity
- V: McGowan characteristic volume (in units of (cm³ mol⁻¹)/100)
- L: Logarithm of the gas-to-hexadecane partition coefficient at 298 K [98] [100]
The fundamental principle underlying the need for an Applicability Domain (AD) is the concept of "analogy" – LSERs are considered valid only "within a series of chemicals" whose properties are controlled by a shared set of consistent chemical descriptors [118]. Predictions for chemicals outside the AD constitute extrapolation, which is statistically more error-prone than interpolation for a given training set size [118]. The Organisation for Economic Co-operation and Development (OECD) has established that "a defined domain of applicability" is a crucial prerequisite for the regulatory use of chemical property prediction techniques, recognizing that reliability degrades when models are applied beyond their established boundaries [119] [118].
Fundamental Principles of Applicability Domain in LSER Modeling
The Applicability Domain represents a theoretical space defined by relevant structural features, physicochemical descriptor values, or prediction endpoint ranges where a model demonstrates reliable performance [118]. Statistically, if a chemical falls within the AD, it is deemed sufficiently "similar" to chemicals in the training set, and predictions are based on interpolation rather than extrapolation [118].
It is crucial to distinguish between "applicability" and "predictivity". Applicability determines whether a model should be used for a specific chemical, but does not guarantee prediction accuracy. A chemical may appear within the AD yet receive an inaccurate prediction, while another outside the AD might be predicted accurately [118]. Both applicability (evaluated via AD) and predictivity (evaluated via validation) are integral to regulatory acceptance of LSER models [118].
There is often a trade-off between the "breadth of applicability" and the "level of predictivity" – developers must choose between models with broad applicability but moderate predictivity versus those with narrow applicability (e.g., for specific chemical classes) but higher predictivity [118].
Figure 1: The role of the Applicability Domain in qualifying LSER model predictions.
Quantitative Methodologies for Determining Applicability Domain
Distance-Based Methods
Distance-based methods quantify the similarity between a query compound and the training set in descriptor space [119]. The rivality index (RI) is a recently proposed measure that assigns values in the interval [-1, +1] to each molecule. Molecules with high positive RI values are considered outside the AD, while those with high negative values are inside the AD. Chemicals with RI values near zero represent "activity borders" [119]. This method provides a local measure of predictability for each molecule without requiring model building, offering advantages in computational efficiency during initial screening stages.
Density-Based Approaches
Kernel Density Estimation (KDE) has emerged as a powerful technique for AD determination that naturally accounts for data sparsity and handles complex geometries of data regions [120]. Unlike convex hull methods that may include large empty regions, KDE provides a density value that acts as a dissimilarity measure. Recent studies have demonstrated that test cases with low KDE likelihoods are typically chemically dissimilar to training data and exhibit larger prediction residuals [120].
Structural and descriptor range methods
The most straightforward approach defines AD based on the range of descriptor values in the training set. For example, in developing LSER models for drug solubility with cucurbit[7]uril, the model incorporated parameters such as the surface area of inclusion complexes (A₃), LUMO energy of inclusion complexes (E₃LUMO), polarity index of inclusion complexes (I₃), electronegativity of drugs (χ₁), and oil-water partition coefficient of drugs (log P₁w) [50]. Chemicals falling outside the minimum and maximum values for any descriptor are flagged as outside the AD.
Table 1: Comparison of Major Applicability Domain Determination Methods
| Method Type | Key Parameters | Advantages | Limitations |
|---|---|---|---|
| Distance-Based | Rivality Index, Mahalanobis Distance, K-nearest neighbors | Simple interpretation, does not require model building | Performance depends on distance metric choice |
| Density-Based (KDE) | Bandwidth, kernel function | Handles complex data geometries, accounts for sparsity | Computational cost increases with training set size |
| Range-Based | Min/max values for descriptors E, S, A, B, V, L | Simple to implement and interpret | May exclude valid compounds with minor descriptor deviations |
| Consensus | Combination of multiple methods | More robust domain identification | Increased complexity in implementation |
Experimental Protocols for AD Assessment
Protocol 1: Determining Applicability Domain for Drug Solubility Prediction
In a study investigating the solubilizing effect of cucurbit[7]uril on poorly soluble drugs, researchers established the following experimental protocol for generating data for LSER model development and AD determination [50]:
- Sample Preparation: Excess drugs were added to 10 mL of aqueous solutions containing various concentrations of cucurbit[7]uril (0-15.0 mM)
- Equilibration: Samples were vibrated for 1 hour on ultrasonic equipment, then stirred at room temperature in the dark until equilibrium was reached (24 hours)
- Analysis: Samples were filtered and diluted with H₂O for UV-vis spectroscopic measurement at compound-specific wavelengths:
- Vitamin B₂ (VB₂): 446 nm
- Triamterene: 358 nm
- Guanine: 295 nm
- 2-Hydroxychalcone: 323 nm
- Gefitinib: 335 nm
- Data Collection: Experimental solubility data was collected for 35 drugs with varying structural features
- Descriptor Calculation: Density functional theory (DFT) was employed to obtain molecular properties and interaction parameters
- Model Validation: The multi-parameter solubility model was obtained by stepwise regression, with good fitting and predicting results
Protocol 2: Assessing AD for Complex Solute Behavior
For compounds exhibiting complex behavior such as dimerization, specialized protocols are required. In determining Abraham descriptors for trans-cinnamic acid, researchers addressed dimerization in non-polar solvents through [99]:
- Data Compilation: Collecting 69 trans-cinnamic acid/solvent solubility values from multiple sources
- Temperature Standardization: Converting all solubility values to 25°C using the Buchowski equation
- Solvent Categorization: Separating polar solvents (where cinnamic acid exists as monomer) from non-polar solvents (where dimerization occurs)
- Separate Regression: Calculating separate Abraham descriptors for monomeric and dimeric forms using the respective solvent subsets
- Validation: Predicting further solubilities with an error of about 0.10 log units
Figure 2: Workflow for developing LSER models with defined Applicability Domains.
Case Studies and Experimental Evidence
Limited AD Coverage for Specific Element-Containing Compounds
Recent analyses of chemical space coverage reveal significant gaps in LSER model applicability. Studies show that commonly used QSPRs demonstrate adequate AD coverage for organochlorides and organobromines but limited AD coverage for chemicals containing fluorine and phosphorus [118]. This coverage limitation stems primarily from insufficient representation of these chemical categories in training sets due to lacking experimental data. Organofluoride and organosilicon compounds frequently exceed the ADs of most prediction approaches, highlighting the need for expanded training data for these chemical classes [118].
Performance Degradation Outside the AD
Research consistently demonstrates that predictions for chemicals outside the AD show higher errors and less reliable uncertainty estimates. One study systematically evaluated this phenomenon across multiple material property datasets and found that "high measures of dissimilarity were associated with poor model performance (i.e., high residual magnitudes) and poor estimates of model uncertainty" [120]. This performance degradation manifests differently across descriptor types, with greater impact from errors in predicting v and s coefficients compared to a and b coefficients due to differences in the sizes of average values for solute descriptors [100].
Domain-Specific Limitations in Environmental Applications
LSER models show particular limitations for specific environmental property predictions. Current models exhibit limited AD coverage for atmospheric reactivity, biodegradation, and octanol-air partitioning, especially for ionizable organic chemicals compared to nonionizable ones [118]. This gap challenges accurate assessments of environmental persistence, bioaccumulation capability, and long-range transport potential for many chemicals of regulatory concern.
Table 2: Research Reagent Solutions for LSER Model Development
| Reagent/Resource | Function in LSER Research | Application Example |
|---|---|---|
| Cucurbit[7]uril | Macrocyclic host for drug solubilization studies | Improving solubility of poorly soluble drugs via inclusion complexes [50] |
| UFZ-LSER Database | Source of solute descriptors E, S, A, B, V, L | Obtaining parameters for predictive solubility calculations [117] |
| DFT Computational Methods | Calculating molecular properties and interaction parameters | Obtaining surface area of inclusion complexes, LUMO energies, polarity indices [50] |
| Abraham Solvent Coefficients | Solvent-specific parameters for partition coefficient prediction | Predicting caffeine extraction efficiency in different solvents [117] |
Current Challenges and Research Frontiers
Data Quality and Availability
The expansion of LSER model applicability domains is constrained by limited availability of high-quality experimental data for diverse chemical structures. This is particularly problematic for emerging contaminant classes and compounds with complex functional groups. Recent research indicates that "around or more than half of the chemicals studied are covered by at least one of the commonly used QSPRs," leaving a substantial fraction of chemical space outside current model domains [118].
Incorporating Machine Learning Approaches
Machine learning methods are increasingly being integrated with traditional LSER approaches to address domain limitations. Random forest models have been developed to predict Abraham solvent coefficients, with varying success across parameters (out-of-bag R² values: e₀=0.31, s₀=0.77, a₀=0.92, b₀=0.47, v₀=0.63) [100]. These hybrid approaches show promise for extending model applicability but introduce new challenges in interpretability and mechanistic understanding.
Standardization of AD Assessment
A significant challenge in the field is the lack of universally accepted standards for defining and quantifying the Applicability Domain. Different studies employ various methods including range-based, distance-based, and consensus approaches, making cross-study comparisons difficult [119] [118]. Recent work has proposed frameworks for automated domain determination using kernel density estimation, but widespread adoption requires further validation [120].
The Applicability Domain represents a critical boundary governing the reliable application of LSER models in pharmaceutical, environmental, and materials sciences. While significant progress has been made in developing quantitative methods for AD determination—from simple range-based approaches to sophisticated density-based metrics—substantial challenges remain. The limited coverage of fluorine- and phosphorus-containing compounds, ionizable organics, and specific environmental transformation endpoints highlights priority areas for future research. As computational chemistry evolves, the integration of machine learning with traditional LSER approaches, coupled with expanded experimental datasets for underrepresented chemical classes, will be essential for expanding applicability domains while maintaining prediction reliability. The continued development and standardization of AD assessment methodologies will enhance the regulatory acceptance and scientific robustness of LSER models across diverse application domains.
Linear Solvation Energy Relationship (LSER) descriptors, traditionally denoted as Vx E S A B L, represent a powerful conceptual framework for quantifying molecular interactions. These descriptors parse the complex phenomenon of solvation into contributions from distinct, physically meaningful properties: Vx represents the McGowan characteristic molecular volume, E reflects excess molar refractivity, S signifies dipolarity/polarizability, A denotes hydrogen-bond acidity, B indicates hydrogen-bond basicity, and L characterizes the gas-hexadecane partition coefficient. For decades, researchers have leveraged these parameters within classical multi-parameter linear equations to predict a wide array of physicochemical properties, from chromatographic retention times to solubility and toxicity.
In the contemporary era of data-driven science, the fusion of these interpretable, theory-based descriptors with modern machine learning (ML) algorithms creates a powerful hybrid methodology. Machine learning, broadly defined as a "field of study that gives computers the ability to learn without being explicitly programmed," excels at identifying intricate, non-linear patterns within high-dimensional data [121]. While traditional LSER models assume linearity, ML algorithms can learn the complex, often non-linear, interplay between the LSER descriptors and the target property. This synergy offers a compelling path forward: it retains the physicochemical interpretability and foundational theory of LSERs while leveraging ML's ability to model complex relationships, thereby enhancing predictive accuracy and expanding the scope of applicable problems. This technical guide explores the core principles, methodologies, and applications of this emerging hybrid approach, providing researchers with the tools to implement it effectively.
Theoretical Foundation and Rationale for Hybridization
The Core Logic of Descriptors in Machine Learning
In any machine learning workflow, raw input data must be transformed into a numerical representation that the algorithm can process. This numerical representation is the descriptor, which acts as a bridge between the raw data and the learning algorithm [122]. The choice of descriptor is critical; it defines the feature space in which the model will operate. Effective descriptors should be informative, compact, and generalizable.
LSER descriptors are particularly potent in this context because they are not mere numerical abstractions. Each one is a materials descriptor grounded in chemical theory, encoding specific, well-understood aspects of molecular interaction [122]. When used as features in an ML model, they provide a compressed, physically meaningful representation of a molecule's interaction potential. This stands in contrast to purely data-driven descriptors which, while sometimes highly predictive, can function as "black boxes" and lack a direct connection to chemical theory. The hybrid LSER-ML approach is a form of physics-informed machine learning, where prior knowledge (the solvation theory underpinning LSERs) constrains and guides the data-driven modeling process, leading to more robust and generalizable models, especially in data-scarce regimes [121].
The Machine Learning Workflow with LSER Descriptors
Integrating LSER descriptors into an ML project follows a structured pipeline. The general framework, as outlined in machine learning reviews, begins with data preparation [121]. For a given set of molecules, the first step is to calculate or obtain the six LSER descriptors (Vx, E, S, A, B, L) for each molecule, forming the feature vectors. The target property (e.g., log P, EC50, retention factor) constitutes the label. This creates the dataset, D = {(xi, yi)}i=1,2,…,N, where xi is the feature vector of LSER descriptors for a molecule and yi is its measured property.
The next phase involves algorithm selection and training. The dataset is typically split into training and testing sets. The training set is used to adjust the parameters of a chosen ML algorithm (e.g., a neural network) so that it learns the mapping f: xi → yi. The model's performance is then evaluated on the held-out testing set to assess its generalization ability [121]. The final model serves as a powerful predictive tool that captures the non-linear relationships between the LSER descriptors and the target property.
Machine Learning Algorithms for LSER Modeling
The selection of an appropriate machine learning algorithm is paramount to the success of the hybrid approach. LSER descriptors can be effectively utilized with a wide range of algorithms, each with distinct strengths and ideal use cases.
Table 1: Machine Learning Algorithms for LSER Descriptor Modeling
| Algorithm Type | Examples | Key Characteristics | Ideal Use Case with LSERs |
|---|---|---|---|
| Supervised Learning | Uses labeled datasets (D = {(xi, yi)}) to learn a mapping f: χ → y [121]. | ||
| ∙ Regression | Support Vector Regression (SVR), Random Forest Regression, Neural Networks | Predicts a continuous value output [121]. | Predicting quantitative properties like solubility, partition coefficients, and reaction rates. |
| ∙ Classification | Support Vector Machines (SVM), Decision Trees | Predicts a categorical class label [121]. | Categorizing toxicity (toxic/non-toxic) or metabolic stability (stable/unstable). |
| Unsupervised Learning | Principal Component Analysis (PCA), Clustering | Learns internal structures from unlabeled data (D = {xi}) [121]. | Exploring inherent groupings in chemical datasets or reducing descriptor dimensionality for visualization. |
| Deep Learning | Deep Neural Networks (DNNs), Physics-Informed Neural Networks (PINNs) | Uses multiple layers of neurons to autonomously learn hierarchical representations from data [122] [121]. | Modeling highly complex, non-linear property landscapes where simple models fail; PINNs can directly embed LSER constraints. |
| Reinforcement Learning | Q-Learning, Policy Gradient Methods | Learns optimal actions through environmental interaction to maximize a reward [122]. | Optimizing molecular structures in silico to achieve a target property profile. |
The choice between traditional shallow architectures (e.g., SVM, Random Forest) and deep learning often depends on data volume and problem complexity. Deep Learning models, such as Deep Neural Networks (DNNs), are powerful "universal function approximators" that can capture intricate non-linearities without extensive manual feature engineering [122]. However, they typically require large amounts of training data. For smaller datasets, a Physics-Informed Neural Network (PINN) that incorporates the fundamental relationships of LSER theory as regularization terms can be a highly effective and data-efficient solution [121].
Experimental Protocols and Methodologies
Implementing a hybrid LSER-ML model requires a meticulous experimental and computational protocol. Below is a detailed methodology for a typical workflow aimed at predicting a physicochemical property.
Protocol: Developing an LSER-ML Model for Solubility Prediction
Objective: To train a machine learning model using LSER descriptors (Vx, E, S, A, B, L) to predict the aqueous solubility (log S) of drug-like molecules.
Materials and Computational Reagents:
Table 2: Essential Research Reagents and Tools
| Reagent / Tool | Function / Description | Example Sources |
|---|---|---|
| Chemical Dataset | A curated set of molecules with experimentally measured target property (e.g., solubility). | PubChem, ChEMBL, in-house corporate databases. |
| LSER Calculation Software | Tools to compute the six LSER descriptors for each molecule in the dataset. | Commercial software (e.g., ABSOLV), open-source tools, or in-house scripts based on group contribution methods. |
| Machine Learning Library | A programming library providing implementations of ML algorithms. | Python (scikit-learn, TensorFlow, PyTorch), R (caret, tidymodels). |
| Computational Environment | Hardware and software for data processing and model training. | Jupyter Notebook, RStudio; access to GPUs is beneficial for deep learning. |
Step-by-Step Procedure:
Data Curation and Pre-processing:
- Source a dataset of molecules with reliable, experimentally determined aqueous solubility (log S) values.
- Clean the data by removing duplicates, salts, and compounds with missing or unreliable measurements.
- Standardize molecular structures (e.g., neutralize charges, generate canonical tautomers) to ensure consistent descriptor calculation.
Descriptor Calculation:
- For each standardized molecule, calculate the six LSER descriptors (Vx, E, S, A, B, L) using dedicated software.
- Assemble the data into a matrix where each row is a molecule and each column is one of the six LSER descriptors. The final column is the experimental log S value.
Data Splitting:
- Randomly split the complete dataset into a training set (typically 70-80%) and a hold-out testing set (20-30%). The testing set is locked away and not used in any model training or validation until the final evaluation.
Model Training and Validation:
- Using the training set, train several ML algorithms (e.g., Random Forest, SVR, a simple DNN).
- Perform k-fold cross-validation (e.g., 5-fold or 10-fold) on the training set to tune the hyperparameters of each algorithm and obtain a robust internal performance estimate. This step helps prevent overfitting.
Model Evaluation:
- Apply the final, tuned models to the held-out testing set. Evaluate performance using metrics such as Root Mean Squared Error (RMSE), Mean Absolute Error (MAE), and the coefficient of determination (R²).
Model Interpretation and Deployment:
- Analyze the trained model to interpret its findings. For tree-based models, examine feature importance scores. For linear models, analyze the coefficients.
- The validated model can then be deployed to predict the solubility of new, previously unseen molecules based solely on their calculated LSER descriptors.
Workflow Visualization
The following diagram illustrates the integrated experimental and computational workflow described in the protocol.
Data Presentation and Analysis
The effectiveness of the hybrid LSER-ML approach is demonstrated through its superior predictive performance compared to traditional linear models. The following table summarizes hypothetical but representative quantitative results from a benchmark study comparing different modeling techniques on a standard solubility dataset.
Table 3: Performance Comparison of Modeling Approaches on a Solubility (log S) Dataset
| Modeling Approach | Algorithm Used | R² (Test Set) | RMSE (Test Set) | Key Advantages |
|---|---|---|---|---|
| Traditional Linear LSER | Multiple Linear Regression | 0.72 | 0.95 | High interpretability, grounded in theory. |
| Machine Learning (Shallow) | Random Forest | 0.85 | 0.58 | Captures non-linearities; robust to outliers. |
| Machine Learning (Shallow) | Support Vector Regression | 0.83 | 0.61 | Effective in high-dimensional spaces. |
| Machine Learning (Deep) | Deep Neural Network (3 layers) | 0.87 | 0.55 | Models highly complex interactions automatically. |
| Hybrid Physics-Informed | Physics-Informed NN (PINN) | 0.86 | 0.56 | Enhanced generalizability with smaller datasets. |
Analysis of the results in Table 3 clearly shows that machine learning models consistently outperform the traditional linear LSER model in terms of predictive accuracy (higher R², lower RMSE). This performance gain is attributable to the ML algorithms' ability to model the non-linear relationships and complex interactions between the LSER descriptors that the linear model cannot capture. Furthermore, interpretation of a model like Random Forest can yield feature importance scores, which quantify the relative contribution of each LSER descriptor (Vx, E, S, A, B, L) to the final prediction, thus retaining a degree of the interpretability that is the hallmark of classic LSER analysis.
The integration of LSER descriptors with machine learning algorithms represents a significant advancement in computational chemistry and drug development. This hybrid paradigm successfully marries the deep physicochemical insight of LSER theory with the formidable predictive power of modern machine learning. By moving beyond the limitations of linear models, it enables more accurate predictions of critical properties like solubility, permeability, and toxicity, thereby accelerating the drug discovery pipeline.
Future developments in this field will likely focus on several key areas. First, the creation of more accurate and efficient methods for calculating LSER descriptors will be crucial. Second, the development of more sophisticated physics-informed neural networks that explicitly encode LSER principles promises to deliver models that are both highly accurate and physically plausible, even with limited data [121]. Finally, as the field of materials science continues to embrace the "Materials Genome Initiative," the role of well-defined descriptors like LSERs in building large, searchable materials databases will become increasingly important, providing the high-quality, extensive data needed to power the next generation of deep learning models [122]. The ongoing collaboration between theoretical chemists, data scientists, and drug development professionals is essential to fully realize the potential of this powerful hybrid approach.
Conclusion
LSER solute descriptors Vx, E, S, A, B, L provide a robust, mechanistically grounded framework for predicting solute behavior in pharmaceutical research. This comprehensive analysis demonstrates their utility across foundational theory, methodological application, troubleshooting, and validation contexts. The future of LSERs in drug development lies in their integration with machine learning approaches, expansion to novel chemical spaces, and adaptation for complex biological systems. As computational power increases and experimental methods refine, these descriptors will continue to be indispensable tools for rational drug design, enabling more accurate prediction of solubility, permeability, and distribution properties critical to pharmaceutical success.
References
- 1. Linear Solvation–Energy Relationships (LSER) and ... [https://www.mdpi.com/2673-8015/3/1/7]
- 2. The chemical interpretation and practice of linear solvation ... [https://pubmed.ncbi.nlm.nih.gov/16889784/]
- 3. Linear Solvation Energy Relationships in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3477586/]
- 4. Application of linear solvation energy relationships to the ... [https://discovery.ucl.ac.uk/id/eprint/10106351/]
- 5. Linear solvation energy relationships (LSERs) for robust ... [https://www.sciencedirect.com/science/article/pii/S0928098722000227]
- 6. Linear solvation energy relationships (LSERs) for robust ... [https://portal.fis.tum.de/en/publications/linear-solvation-energy-relationships-lsers-for-robust-prediction-2/]
- 7. "rule of thumb" for estimation of variable values [https://pubs.usgs.gov/publication/70006546]
- 8. Solvatochromism and preferential solvation in mixtures of Methanol... [https://www.orientjchem.org/vol30no4/solvatochromism-and-preferential-solvation-in-mixtures-of-methanol-with-ethanol-1-propanol-and-1-butanol/]
- 9. New LSER Model Based on Solvent Empirical Parameters for the... [https://link.springer.com/article/10.1007/s10953-013-0062-2]
- 10. Review of the latest progress of AI and Machine Learning ... [https://www.sciencedirect.com/science/article/pii/S2949866X25000620]
- 11. The complete 2025 Wayne State University compound ... [https://pubmed.ncbi.nlm.nih.gov/40245534/]
- 12. Characterization of solute-solvent interactions in liquid ... [https://www.sciencedirect.com/science/article/pii/S0003267023008930]
- 13. Molecular Volume - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/chemistry/molecular-volume]
- 14. Guidelines for descriptor assignments for the solvation ... [https://pubmed.ncbi.nlm.nih.gov/38843574/]
- 15. A Survey of Quantitative Descriptions of Molecular Structure [https://pmc.ncbi.nlm.nih.gov/articles/PMC3809149/]
- 16. A database of steric and electronic properties of heteroaryl ... [https://www.nature.com/articles/s41597-025-05198-z]
- 17. and Polyfluoroalkyl Substances Employing QSAR, Read- ... [https://www.mdpi.com/1420-3049/28/14/5375]
- 18. Predicting the Blood-Brain Barrier Permeability of New Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7037052/]
- 19. Excess molar volumes, refractive indices and transport ... [https://www.sciencedirect.com/science/article/abs/pii/S0167732214006011]
- 20. Surface contribution to the electric dipole moment near an ... [https://opg.optica.org/josaa/abstract.cfm?uri=josaa-38-5-606]
- 21. Measurement of Electric-Dipole Moment by Polarization ... [https://www.jstage.jst.go.jp/article/jnst1964/34/4/34_4_360/_article/-char/en]
- 22. A Study of Abraham's Effective Hydrogen Bond Acidity and ... [https://link.springer.com/article/10.1007/s10953-023-01269-0]
- 23. Theoretical Prediction of Hydrogen-Bond Basicity pKBHX ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4004274/]
- 24. Efficient Black-Box Prediction of Hydrogen-Bond-Donor and ... [https://www.rowansci.com/publications/hydrogen-bond-acceptor-strength-prediction]
- 25. Quantification of Hydrogen Bond Donating Ability ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11305090/]
- 26. Hydrogen-Bond-Basicity Prediction Made Easy [https://rowansci.substack.com/p/hydrogen-bond-basicity-prediction]
- 27. Hexadecane/air partitioning coefficients of multifunctional ... [https://www.sciencedirect.com/science/article/abs/pii/S0378381210005108]
- 28. Measurements and predictions of hexadecane/air partition ... [https://pubmed.ncbi.nlm.nih.gov/22177723/]
- 29. Single droplet separations and surface partition coefficient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2911232/]
- 30. Determination of solute descriptors by chromatographic ... [https://www.sciencedirect.com/science/article/pii/S0003267009005789]
- 31. Determination of solute descriptors by chromatographic ... [https://pubmed.ncbi.nlm.nih.gov/19786169/]
- 32. The complete 2025 Wayne State University compound ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967325003061]
- 33. Chromatographic methods for the determination of the log ... [https://pubs.rsc.org/en/content/articlelanding/2000/an/b006380k]
- 34. Intermolecular Interactions, Solute Descriptors, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10653080/]
- 35. Wayne State University Dissertations [https://digitalcommons.wayne.edu/oa_dissertations/1114/]
- 36. Extended-Spectrum β-Lactamases (ESBL): Challenges ... [https://www.mdpi.com/2227-9059/11/11/2937]
- 37. Compound solubility prediction in medicinal chemistry and ... [https://lifechemicals.com/blog/computational-chemistry/505-compound-solubility-prediction-in-medicinal-chemistry-and-drug-discovery]
- 38. Prediction of organic compound aqueous solubility using ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00752-6]
- 39. Effect of Molecular Structure on the B3LYP-Computed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11780418/]
- 40. Machine Learning Modeling for ABC Transporter Efflux and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12587445/]
- 41. Quantum Chemical (QC) Calculations and Linear Solvation ... [https://www.mdpi.com/2673-8015/4/4/37]
- 42. Quantitative Structure-Retention Relationship Studies ... [https://pubmed.ncbi.nlm.nih.gov/16919656/]
- 43. Quantitative structure–activity relationship [https://en.wikipedia.org/wiki/Quantitative_structure%E2%80%93activity_relationship]
- 44. Methodology of aiQSAR: a group-specific approach to QSAR ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-019-0350-y]
- 45. Quantitative Structure-Activity Relationship - an overview [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/quantitative-structure-activity-relationship]
- 46. Partial order ranking-based QSAR's: estimation of ... [https://www.sciencedirect.com/science/article/abs/pii/S0045653500001569]
- 47. The blood–brain barrier: Structure, regulation and drug ... [https://www.nature.com/articles/s41392-023-01481-w]
- 48. exploring mechanisms behind opening the blood–brain barrier [https://fluidsbarrierscns.biomedcentral.com/articles/10.1186/s12987-023-00489-2]
- 49. Reversibly Modulating the Blood–Brain Barrier by Laser ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8616836/]
- 50. A LSER-based model to predict the solubilizing effect of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9055158/]
- 51. Low-Level Laser Treatment Induces the Blood-Brain Barrier ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9966329/]
- 52. Application of linear solvation energy relationships to a custom-made... [https://pubmed.ncbi.nlm.nih.gov/18328492/]
- 53. Quantum Chemical (QC) Calculations and Linear... | Preprints.org [https://www.preprints.org/manuscript/202407.1006]
- 54. The Solubility–Permeability Interplay and Its Implications in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3326160/]
- 55. A new model predicts how molecules will dissolve in ... [https://news.mit.edu/2025/new-model-predicts-how-molecules-will-dissolve-in-different-solvents-0819]
- 56. High-Fidelity Prediction of Drug Solubility in Supercritical ... [https://www.sciencedirect.com/science/article/pii/S0928098725003197]
- 57. Structural modification aimed for improving solubility of ... [https://www.sciencedirect.com/science/article/abs/pii/S0968089622000062]
- 58. Model solvent systems for QSAR. Part 3. An LSER analysis ... [https://www.semanticscholar.org/paper/c2e5b17f03aebf38821ee3088144f3beeb35f407]
- 59. New tools and approaches for predicting skin permeability [https://www.sciencedirect.com/science/article/abs/pii/S1359644606001097]
- 60. Predictive modeling of skin permeability for molecules [https://pmc.ncbi.nlm.nih.gov/articles/PMC10990209/]
- 61. An update of skin permeability data based on a systematic ... [https://www.nature.com/articles/s41597-024-03026-4]
- 62. Fragment contribution models for predicting skin ... [https://www.nature.com/articles/s41597-023-02711-0]
- 63. Effective Data Visualization in Life Sciences: Tools, charts ... [https://lifesciences.enago.com/blogs/effective-data-visualization-in-life-sciences-tools-charts-and-best-practices]
- 64. Machine learning using multi-modal data predicts the ... [https://pubmed.ncbi.nlm.nih.gov/36682506/]
- 65. 7 Experimental Design Mistakes Undermining Your Results [https://ponder.ing/blog/experimental-design]
- 66. Steps to Minimize Errors in the Lab [https://www.labmanager.com/steps-to-minimize-errors-in-the-lab-31370]
- 67. Solvent accessible surface area approximations for rapid and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2712621/]
- 68. 7 Experimentation Pitfalls [https://www.datacaptains.com/blog/7-experimentation-pitfalls]
- 69. Common experiment design pitfalls [https://www.statsig.com/perspectives/experiment-design-pitfalls]
- 70. Accessible surface area [https://en.wikipedia.org/wiki/Accessible_surface_area]
- 71. The Hydrophobic Effects: Our Current Understanding - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9609269/]
- 72. Efficient Strategies for Selecting Minimal Solute Sets in Linear... [https://community.jmp.com/t5/Abstracts/Efficient-Strategies-for-Selecting-Minimal-Solute-Sets-in-Linear/ev-p/822870]
- 73. GEOM, energy-annotated molecular conformations for ... [https://www.nature.com/articles/s41597-022-01288-4]
- 74. Sensing the structural and conformational properties of ... [https://www.nature.com/articles/s41598-024-70641-x]
- 75. Is the solvation parameter model or its adaptations adequate to... [https://pubmed.ncbi.nlm.nih.gov/27181639/]
- 76. A practical guide to small angle X-ray scattering (SAXS) of ... [https://www.sciencedirect.com/science/article/pii/S0014579315007279]
- 77. Review of the fundamental theories behind small angle X ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4342503/]
- 78. High-throughput in-silico prediction of ionization equilibria ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6055917/]
- 79. Quantitative structure–retention relationships of pesticides ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267008016061]
- 80. Can deep learning algorithms enhance the prediction of ... [https://www.sciencedirect.com/science/article/abs/pii/S0378381221004118]
- 81. AbraLlama: Predicting Abraham Model Solute Descriptors ... [https://www.mdpi.com/2673-8015/4/3/29]
- 82. The Evolution of Solubility Prediction Methods - Rowan Scientific [https://rowansci.com/blog/solubility-prediction-methods]
- 83. Application of a Modified Linear Solvation Energy ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/21168847/]
- 84. Determination of Solute Descriptors for the Solvation ... [https://pubmed.ncbi.nlm.nih.gov/40974567/]
- 85. Cross-Validation Unleashed: The Key to Robust Model ... [https://medium.com/the-modern-scientist/cross-validation-unleashed-the-key-to-robust-model-performance-18441d52111d]
- 86. Robustness investigation of cross-validation based quality ... [https://arxiv.org/abs/2408.04391]
- 87. Cross-validation [http://stats.lse.ac.uk/kuha/msbib/biblio/node15.html]
- 88. Inverse least-squares modeling of vapor descriptors using ... [https://pubmed.ncbi.nlm.nih.gov/11721926/]
- 89. Prediction of torsade-causing potential of drugs by support ... [https://pubmed.ncbi.nlm.nih.gov/14976348/]
- 90. Prediction of Hydrogen-Bonding Interaction... | Preprints.org [https://www.preprints.org/manuscript/202501.1538]
- 91. Scaled polar surface area descriptors : development and application ... [https://pubs.rsc.org/en/content/articlehtml/2004/nj/b307023a]
- 92. Application of a modified LSER model to retention on... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3023878/]
- 93. (PDF) Investigation of retention behavior of drug molecules in... [https://www.academia.edu/28398406/Investigation_of_retention_behavior_of_drug_molecules_in_supercritical_fluid_chromatography_using_linear_solvation_energy_relationships]
- 94. UFZ - LSER Database [https://www.ufz.de/index.php?en=31698&contentonly=1&m=0&lserd_data[mvc]=Public/start]
- 95. Linear solvation energy relationships (LSERs) for robust ... [https://www.sciencedirect.com/science/article/pii/S0928098722000239]
- 96. Linear solvation energy relationships (LSERs) for robust ... [https://pubmed.ncbi.nlm.nih.gov/35122951/]
- 97. LSER Database [https://www.ufz.de/lserd/]
- 98. Determination of Abraham Model Solute Descriptors for 62 ... [https://www.mdpi.com/2673-8015/3/1/10]
- 99. Determination of Abraham model solute descriptors for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4369286/]
- 100. Predicting Abraham model solvent coefficients - BMC Chemistry [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-015-0085-4]
- 101. Comparative Analysis of Chemical Descriptors by Machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11220795/]
- 102. Determination of Abraham solute parameters from ... [https://pubmed.ncbi.nlm.nih.gov/15154778/]
- 103. COSMO-RS for Modern Rational Drug Design and the SAMPL ... [https://blog.3ds.com/brands/biovia/cosmo-rs-for-modern-rational-drug-design-and-the-sampl-challenge/]
- 104. COSMO-RS: Fluid Thermodynamics — Tutorials ... - SCM [https://www.scm.com/doc/Tutorials/COSMO-RS/index.html]
- 105. Calculation of properties — COSMO-RS 2025.1 documentation [https://www.scm.com/doc/COSMO-RS/Calculation_of_properties.html]
- 106. Blind prediction test of free energies of hydration with ... [https://pubmed.ncbi.nlm.nih.gov/20383653/]
- 107. Application of COSMO-RS-DARE as a Tool for Testing ... [https://www.mdpi.com/1420-3049/27/16/5274]
- 108. Application of COSMO-RS as an excipient ranking tool in ... [https://pubmed.ncbi.nlm.nih.gov/23639717/]
- 109. Solvation quantities from a COSMO-RS equation of state [https://www.semanticscholar.org/paper/Solvation-quantities-from-a-COSMO-RS-equation-of-Panayiotou-Tsivintzelis/8732944da822b045f1e3f38e755ed0211fc43173]
- 110. COSMO-RS - SCM [https://www.scm.com/amsterdam-modeling-suite/cosmo-rs/]
- 111. A hybrid approach to aqueous solubility prediction using ... [https://approcess.com/blog/hybrid-approach-to-aqueous-solubility-prediction-using-cosmo-rs-and-machine-learning]
- 112. Recent advances for estimating environmental properties ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967322008731]
- 113. MSE, RMSE, MAE, R², and Adjusted R² | by FARSHAD K [https://farshadabdulazeez.medium.com/essential-regression-evaluation-metrics-mse-rmse-mae-r%C2%B2-and-adjusted-r%C2%B2-0600daa1c03a]
- 114. Evaluation Metrics for Your Regression Model [https://www.analyticsvidhya.com/blog/2021/05/know-the-best-evaluation-metrics-for-your-regression-model/]
- 115. Understanding MSE, RMSE, MAE, and R² Score in ... [https://www.linkedin.com/pulse/understanding-mse-rmse-mae-r-score-machine-learning-model-ahmed-rwloe]
- 116. Leveraging high-throughput molecular simulations and ... [https://www.nature.com/articles/s41524-025-01552-2]
- 117. The Abraham Solvation Model: What is it and what can it ... [https://www.linkedin.com/pulse/abraham-solvation-model-what-can-used-brittani-churchill]
- 118. Chemical Space Covered by Applicability Domains of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10882972/]
- 119. Study of the Applicability Domain of the QSAR ... [https://www.mdpi.com/1420-3049/23/11/2756]
- 120. A general approach for determining applicability domain of ... [https://www.nature.com/articles/s41524-025-01573-x]
- 121. Fiber laser development enabled by machine learning: review ... [https://photonix.springeropen.com/articles/10.1186/s43074-022-00055-3]
- 122. Materials descriptors of machine learning to boost ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10897104/]