Batch vs. Continuous Manufacturing: A Detailed PMI Comparison for Sustainable Pharma
This article provides a comprehensive analysis of Process Mass Intensity (PMI) in batch versus continuous pharmaceutical manufacturing.
Batch vs. Continuous Manufacturing: A Detailed PMI Comparison for Sustainable Pharma
Abstract
This article provides a comprehensive analysis of Process Mass Intensity (PMI) in batch versus continuous pharmaceutical manufacturing. Tailored for researchers, scientists, and drug development professionals, it explores the foundational principles of PMI, methodologies for its calculation and application, strategies for troubleshooting and optimization, and a final validation through direct technical and economic comparison. The synthesis aims to serve as a decision-making tool for adopting more sustainable and efficient production processes.
Understanding Process Mass Intensity (PMI) in Pharmaceutical Manufacturing
Process Mass Intensity (PMI) is a key green chemistry metric used to provide a holistic assessment of the mass efficiency of a manufacturing process. It is defined as the total mass of materials used (including raw materials, reactants, and solvents) to produce a specified mass of product, making it an indispensable indicator of a process's environmental footprint and resource efficiency [1].
Frequently Asked Questions (FAQs)
What does PMI measure? PMI measures the total mass of all materials (raw materials, reactants, and solvents) required to manufacture a unit mass of a desired product, typically expressed as kg of material per kg of Active Pharmaceutical Ingredient (API) [1].
Why is a lower PMI value desirable? A lower PMI indicates a more efficient and environmentally friendly process, as it signifies less material consumption and, consequently, less waste generated. This translates to reduced environmental impact and lower costs [1].
How does PMI differ from simple chemical yield? Unlike chemical yield, which only measures the efficiency of converting reactants to product, PMI provides a more comprehensive view by including all materials used in the process, such as solvents for reactions, purification, and isolation [1].
What are typical PMI values in the pharmaceutical industry? PMI varies significantly by therapeutic modality. Small molecule APIs have the lowest PMI (median 168-308), followed by biopharmaceuticals (average ~8,300), with synthetic peptides having the highest PMI (average ~13,000 for SPPS) [1].
In a continuous process, is PMI the only metric to consider for sustainability? No. For biologics, studies show continuous processes can have a PMI similar to batch processes. However, because continuous processes can produce more product per unit of time, they may consume less energy per kg of drug substance, leading to better overall environmental sustainability that isn't captured by PMI alone [2].
Troubleshooting Guide: High PMI in Peptide Synthesis
Solid-phase peptide synthesis (SPPS) is a common but resource-intensive process. The following guide addresses common issues leading to high PMI.
| Problem Area | Specific Issue | Potential Impact on PMI | Recommended Solution |
|---|---|---|---|
| Solvent Usage | Use of large excesses of solvents like DMF, DMAc, NMP, DCM, and TFA for coupling, washing, and cleavage [1]. | Major contributor to high PMI. | Optimize solvent volumes per cycle, investigate solvent recycling where possible, and research alternative, greener solvents [1]. |
| Reagent Efficiency | Use of large excesses of amino acids and coupling reagents to drive reactions to completion [1]. | Increases mass of raw materials. | Optimize coupling reagent and protected amino acid stoichiometry. Use real-time monitoring to ensure completion without excessive overuse [1]. |
| Process Design | Inefficient isolation and purification methods (e.g., lyophilization, chromatography) requiring high solvent volumes [1]. | Significant waste in downstream steps. | Explore more efficient purification techniques and optimize isolation protocols to reduce solvent consumption post-synthesis [1]. |
| Technology Choice | Reliance solely on SPPS for longer peptides where yields decrease with length [1]. | Leads to high material use and poor overall yield. | For longer peptides, consider hybrid approaches (e.g., SPPS of fragments followed by liquid-phase fragment coupling) or alternative technologies like recombinant synthesis [1]. |
Experimental Protocol: Calculating and Comparing PMI
This protocol provides a standardized methodology for calculating the PMI of a process, enabling a direct comparison between batch and continuous manufacturing.
1. Objective To quantitatively determine the Process Mass Intensity (PMI) for a given API synthesis process and use this data to compare the material efficiency of different manufacturing approaches.
2. Materials and Data Collection
- Data Source: Detailed batch production records or process flow sheets.
- Materials to Tally: The total mass of every input entering the process must be recorded. This includes:
- All reactants and reagents.
- All solvents (used in reactions, work-ups, crystallizations, and purifications).
- Catalysts.
- Purification materials (e.g., chromatography resins, filter aids).
- Note: Water is typically excluded from the PMI calculation [1].
- Output: The total mass of the final, purified Active Pharmaceutical Ingredient (API) produced.
3. Calculation
Use the following formula to calculate PMI:
PMI (kg/kg) = Total Mass of All Input Materials (kg) / Total Mass of API Produced (kg)
4. Comparative Analysis: Batch vs. Continuous
- Once the PMI for both a batch process and a comparable continuous process is calculated, the values can be directly compared.
- Interpretation: A lower PMI indicates superior material efficiency.
- Advanced Analysis: To gain a fuller sustainability picture, complement the PMI data with an assessment of total energy consumption per kg of API produced, as continuous processes may offer advantages not reflected in PMI alone [2].
PMI Data for Different Pharmaceutical Modalities
The table below summarizes typical PMI values across different drug modalities, providing a benchmark for evaluating your own processes [1].
| Therapeutic Modality | Typical PMI (kg/kg API) | Key Context |
|---|---|---|
| Small Molecule APIs | Median: 168 - 308 | Represents the benchmark for high material efficiency. |
| Oligonucleotides | Average: ~4,299 | Also synthesized via solid-phase support, leading to higher PMI. |
| Biopharmaceuticals | Average: ~8,300 | Includes monoclonal antibodies and other large biologics. |
| Synthetic Peptides (SPPS) | Average: ~13,000 | The highest PMI of the group, driven by solvent and reagent excess. |
The Scientist's Toolkit: Key Reagents & Materials
The following table details critical materials used in peptide synthesis, a field where PMI optimization is a significant focus [1].
| Research Reagent / Material | Function in the Experiment / Process |
|---|---|
| Fmoc-Protected Amino Acids | Building blocks for constructing the peptide chain in SPPS; the Fmoc group prevents unwanted reactions [1]. |
| Resin Solid Support | An insoluble polymer bead that serves as the anchor point for the growing peptide chain during SPPS, enabling the use of excess reagents [1]. |
| Coupling Reagents (e.g., HATU, DIC) | Activates the carboxylic acid of the incoming amino acid, facilitating bond formation with the growing peptide chain [1]. |
| Solvents (DMF, NMP, DCM) | Swells the resin and serves as the reaction medium; a major contributor to PMI [1]. |
| Cleavage Cocktail (e.g., TFA) | A strong acid mixture used to cleave the finished peptide from the solid resin and remove protecting groups [1]. |
Workflow Diagram: PMI Calculation & Comparison
The diagram below outlines the logical workflow for calculating PMI and performing a batch-versus-continuous comparison.
Comparative Analysis Logic
This diagram illustrates the key decision-making process when analyzing PMI data between two manufacturing processes.
Core Principles of Batch Process Manufacturing
Batch process manufacturing is a production method where groups of identical products are created together in specific quantities, or batches. Each batch completes its stage of the manufacturing process before the entire group moves to the next phase [3]. This approach is particularly valuable in industries like pharmaceuticals, where precision, customization, and quality control are paramount [4] [5].
Unlike continuous manufacturing, which involves an uninterrupted production flow, batch processing allows for adjustments between production runs, making it ideal for research and development, small-scale production, and products requiring high levels of customization [6]. This technical support center provides troubleshooting guidance and foundational knowledge for scientists and drug development professionals working with batch processes.
Key Characteristics and Comparison
Batch manufacturing is defined by several core principles that distinguish it from other production methods, particularly continuous manufacturing.
Defining Characteristics
- Group Production: Items are produced in specified groups or batches, with all units in a batch undergoing each processing step together before moving to the next stage [5] [7].
- Flexibility: Equipment can be reconfigured between batches to produce different product variations, allowing manufacturers to adapt to changing demands and produce diverse product lines [4] [3].
- Quality Control: Products are examined at multiple stages throughout the production process, enabling improved monitoring and quality assurance [5].
- Distinct Batches: Each production batch is separate and identifiable, facilitating traceability through lot numbers for recalls and compliance purposes [3].
Batch vs. Continuous Manufacturing
The table below summarizes the key differences between batch and continuous processes, which is particularly relevant for pharmaceutical manufacturing research and development.
| Characteristic | Batch Process | Continuous Process |
|---|---|---|
| Production Flow | Defined start and end points; sequential processing [6] | Ongoing, uninterrupted manufacturing with single production line [6] |
| Volume | Suitable for small to medium volumes [4] | Designed for large-scale, high-volume output [4] |
| Flexibility | High; allows adjustments between batches [4] | Low; designed for specific product type [4] |
| Quality Control | End-of-process checks and adjustments based on previous batches [4] | Real-time monitoring with automated systems for immediate corrections [4] |
| Cost Structure | Lower initial investment, higher per-unit costs [4] | High initial investment, lower operational and per-unit costs [4] |
| Equipment | Simpler, more versatile equipment [4] | Sophisticated, specialized equipment for prolonged operation [4] |
| Ideal Application | Customization, varied products, R&D [5] | Standardized, high-volume production [4] |
Batch Manufacturing QA Workflow
Advantages and Disadvantages
Understanding the trade-offs of batch process manufacturing is essential for effective experimental design and troubleshooting.
Advantages
- Cost Efficiency for Moderate Volumes: Reduced expenses through bulk material purchasing and repeated equipment use, distributing setup costs across multiple batches [3] [5].
- Enhanced Quality Control: Improved monitoring at various production stages allows for detection and correction of issues before batch completion [5] [7].
- Production Flexibility: Ability to produce different product variations using the same equipment, enabling adaptation to seasonal demand or research requirements [3] [5].
- Reduced Waste: Producing aligned with actual demand helps prevent surplus products and minimizes waste [3].
- Improved Traceability: Distinct batch identification facilitates tracking for recalls, compliance, and research replication [3].
Disadvantages
- Extended Production Times: Sequential production phases and setup between batches can extend overall manufacturing time compared to continuous methods [3].
- Higher Per-Unit Costs: More manufacturing stages and less efficient equipment use typically result in higher costs than mass production [3] [7].
- Risk of Complete Batch Failure: A single error can compromise an entire batch, resulting in significant material and time losses [3].
- Increased Labor Requirements: Frequent setup and adjustments between batches often require more skilled labor and specialized attention [3] [5].
Common Batch Processing Issues and Troubleshooting
The following section addresses frequent challenges in batch processing environments and provides methodological guidance for resolution.
FAQ: Batch Job Results in Error
Q: A batch job has failed during pharmaceutical formulation. What systematic approach should I take to diagnose the issue?
Methodology for Diagnosis:
- Review Batch Run Tree: Examine the batch execution hierarchy to identify which specific thread or process failed. Expand all tree levels to view thread status and error messages [8].
- Analyze Log Files: Download and review both stdout and stderr log files. Search for keywords like "error" or database error codes (e.g., "ORA-") to identify specific failure points [8].
- Resubmit with Single Thread: For complex errors, resubmit the batch job with a single thread and the "Max-Errors" parameter set to a low value (e.g., 10) to isolate the issue without processing excessive errors [8].
- Verify Data Quality: For database-related errors, check for data integrity issues including foreign key validation and statistical anomalies in loaded data [8].
- Check Resource Allocation: Confirm that thread pool workers are available and not exhausted by other concurrent processes [8].
Preventive Measures:
- Implement data validation batch programs before full processing [8]
- Establish baseline performance metrics with smaller data volumes [8]
- Regularly monitor database index status and query performance [8]
FAQ: Batch Performance Degradation
Q: My previously stable batch process has developed significant performance issues. What factors should I investigate?
Experimental Protocol for Performance Analysis:
- Concurrent Process Audit: Document all activities running concurrently with the batch process, including other batch jobs, system maintenance, or data conversion activities [8].
- Database Performance Analysis:
- Execute index validation query:
SELECT INDEX_NAME, TABLE_NAME FROM all_indexes where TABLE_OWNER = 'CISADM' and STATUS = 'UNUSABLE' ORDER BY TABLE_NAME[8] - Monitor database resource utilization during batch execution
- Execute index validation query:
- Multi-threading Optimization:
- Establish baseline with reduced thread count
- Incrementally increase threads while measuring performance impact
- Note that doubling threads does not typically halve processing time [8]
- Extension Impact Assessment: Review any custom scripts or algorithms for inefficient SQL or resource-intensive operations [8].
FAQ: Data Integrity and Quality Issues
Q: My batch process completes but produces inconsistent or invalid data outputs. How can I identify the root cause?
Diagnostic Workflow:
- Schema Validation: Confirm all data inputs comply with expected schemas and that schema evolution hasn't introduced compatibility issues [9].
- Data Lineage Tracing: Implement data provenance tracking to identify where transformations introduce inconsistencies [9].
- Statistical Sampling: Utilize validation batch programs to perform statistical sampling of processed data and identify patterns in errors [8].
- Real-time Monitoring Implementation: For critical processes, implement process analytical technology (PAT) tools to monitor quality attributes during processing rather than only at completion [6].
Batch Process Troubleshooting Logic
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key materials and their functions in pharmaceutical batch process research and development.
| Material/Reagent | Function in Batch Process Research |
|---|---|
| Process Analytical Technology (PAT) Tools | Enable real-time monitoring of critical quality attributes during batch processing [6] |
| Excipients with Specific Flow Profiles | Ensure proper powder blending and homogeneity in solid dosage batch production [6] |
| Reference Standards | Provide quality control benchmarks for evaluating batch consistency and compliance [5] |
| Specialized Sensor Arrays | Monitor process parameters (temperature, pressure, pH) throughout batch cycles [6] |
| Traceability Markers | Facilitate batch tracking and identification throughout the production lifecycle [3] |
| Cleaning Validation Agents | Verify equipment cleanliness between different product batches to prevent cross-contamination [4] |
| Stability Testing Materials | Assess shelf-life and degradation profiles of batch-produced pharmaceuticals [5] |
Best Practices for Batch Process Optimization
Implementation of Batch Tracking Systems
Batch tracking technology connects with machinery to enhance monitoring and control of production. Effective implementation includes [5]:
- Assigning unique, traceable identification to each produced batch
- Including expiration dates for perishable items
- Establishing connections between raw materials and final products
- Enabling rapid response to quality issues or recalls
Production Schedule Optimization
Effective scheduling is essential for efficient batch manufacturing [7]:
- Align production with sales patterns to reduce overproduction risk
- Implement capacity planning to determine needed production capacity
- Utilize automation for demand forecasting and schedule adjustments
- Schedule maintenance during natural downtimes between batches
Quality by Design (QbD) Principles
Implement QbD approaches to enhance batch consistency [6]:
- Define critical process parameters and their acceptable ranges
- Establish design space for flexible operation within validated parameters
- Implement control strategies based on risk assessment
- Utilize statistical process control for continuous monitoring
Core Principles of Continuous Process Manufacturing
Continuous process manufacturing represents a fundamental shift in production philosophy, particularly for pharmaceutical researchers and drug development professionals. Unlike traditional batch processing, where materials move in discrete groups through separate operations, continuous manufacturing involves the uninterrupted flow of materials through an integrated system [6]. This approach offers significant advantages in speed, quality control, and efficiency but requires deep understanding of its core principles for successful implementation and troubleshooting [10] [11].
This technical support center provides practical guidance structured within the broader context of batch versus continuous pharmaceutical manufacturing comparison research. The following sections address frequently asked questions, troubleshooting guides, and experimental protocols to support scientists navigating the transition to continuous systems.
Core Principles and Definitions
What is Continuous Process Manufacturing?
Continuous process manufacturing is a production approach where material inputs and outputs occur simultaneously throughout the process without intermittent pauses or human involvement between the beginning and end of production [11]. This creates a steady stream of output where products advance from one stage to the next immediately upon completion [12].
The Five Key Principles of Continuous Flow
Implementing successful continuous flow manufacturing requires adherence to five essential principles that work together to create efficient production systems:
- Takt Time Alignment: The production rate is calibrated to customer demand to avoid overproduction and idle time [10].
- Standardized Work: Each task is performed consistently every time to reduce variation and maintain quality [10].
- Minimized Work-in-Progress (WIP): Keeping work-in-progress to a minimum allows issues to surface more quickly and prevents buildup between process steps [10].
- Balanced Workload (Heijunka): Workloads are distributed evenly across workstations to avoid bottlenecks and overburdening [10].
- Reliable, Flexible Processes: Processes must be stable enough to avoid breakdowns yet adaptable to demand shifts [10].
Comparison with Batch Manufacturing
The table below summarizes key quantitative differences between batch and continuous manufacturing approaches:
| Aspect | Continuous Flow Manufacturing | Batch Manufacturing |
|---|---|---|
| Production Speed | Constant output [12] | Intermittent output [12] |
| Lead Time Reduction | 50-70% reduction [12] | No significant reduction |
| Quality Control | Real-time monitoring [12] | End-of-batch testing [12] |
| Defect Rate Reduction | Up to 90% [12] | Limited reduction |
| Inventory Costs | 30-50% reduction [12] | Higher inventory expenses [12] |
| Variable Cost Reduction | 40-50% for pharmaceuticals [11] | Higher per-unit costs [12] |
| Production Flexibility | Limited product variation capabilities [12] | Easy product changeovers [12] |
| Initial Investment | Higher [12] [11] | Lower [12] |
Five Lean Principles Workflow
Troubleshooting Guides
FAQ: Common Operational Challenges
Q: What are the most frequent barriers to implementing continuous flow in pharmaceutical manufacturing?
A: Common barriers include:
- High setup times that make large batches tempting [10]
- Process variability in cycle times or quality that breaks flow quickly [10]
- Equipment limitations, as flow reactor pipes may be ineffective for some APIs and solids can cause blockages [11]
- Organizational resistance due to high startup costs ($4-5 million for new equipment) and comfort with established batch methods [11]
Q: How does continuous manufacturing improve quality control compared to batch systems?
A: Continuous systems enable real-time quality assurance through constant monitoring, allowing immediate detection and correction of issues rather than discovering defects after completing entire batches [12] [11]. This results in up to 90% reduction in defect rates and minimizes large-scale product rejection [12].
Q: What analytical approaches are recommended for troubleshooting particle contamination?
A: For particle contamination issues, employ a tiered analytical strategy [13]:
- First step: Physical methods like scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDX) for chemical identification of inorganic compounds
- Second step: Chemical approaches including qualitative solubility tests and chromatographic methods (LC-HRMS, GC-MS) for structure elucidation
- Advanced techniques: Nuclear magnetic resonance (NMR) for complex contaminant identification
Troubleshooting Specific Issues
Problem: Flow Interruptions and Bottlenecks
Symptoms: Irregular output, accumulation of work-in-progress between stations, delayed cycle times
Root Cause Analysis Methodology:
- Map the current process using value stream mapping to visualize workflow and identify delay points [10]
- Measure cycle times at each station to identify imbalances
- Check equipment reliability data for frequent breakdowns
- Verify material quality and consistency from suppliers
Corrective Actions:
- Balance workloads using Heijunka (production leveling) principles [10]
- Implement predictive maintenance schedules for critical equipment
- Cross-train employees to increase flexibility and address staffing imbalances [10]
- Redesign factory layout to minimize transport distances and enable smooth material flow [12]
Problem: Quality Variations in Final Product
Symptoms: Inconsistent potency, contamination events, failed quality control checks
Root Cause Analysis Methodology:
- Review Process Analytical Technology (PAT) data for correlation with process parameters [6]
- Analyze trend data from real-time monitoring systems [12]
- Conduct material traceability assessment throughout the production stream
- Perform equipment calibration verification on critical control points
Corrective Actions:
- Enhance real-time monitoring with additional PAT tools at identified vulnerability points [6]
- Strengthen supplier qualification processes for raw materials [14]
- Implement automated quality checks throughout the production line [12]
- Establish tighter control strategies for critical process parameters [6]
Experimental Protocols and Methodologies
Protocol: Transitioning from Batch to Continuous Manufacturing
Purpose: Systematically evaluate and implement continuous manufacturing for a pharmaceutical product currently produced using batch methods.
Workflow:
Batch to Continuous Transition Workflow
Methodology Details:
Assess Current Process Flows [12]
- Document production rates, cycle times, and equipment utilization
- Identify bottlenecks, delays, and quality control points
- Establish baseline metrics for measuring improvements
Identify and Map Value Streams [12]
- Track material movement paths and information flow
- Identify non-value-adding activities using Lean principles [15]
- Measure process timing and locate improvement opportunities
Design Optimized Factory Layout [12]
- Minimize transport distances between equipment
- Enable smooth material flow with reduced operator movement
- Optimize space utilization with ergonomic workstation design
Develop Employee Training Program [12]
- Cover continuous flow fundamentals and quality control procedures
- Provide technical skills for equipment operation and monitoring
- Implement problem-solving methods and standard work practices
Protocol: Root Cause Analysis for Quality Defects
Purpose: Systematically investigate and resolve quality issues in continuous manufacturing processes to prevent recurrence.
Methodology:
Information Collection [13]
- Document what happened with detailed problem description
- Establish when the incident occurred (time frame)
- Identify who was involved (personnel, materials, equipment)
Analytical Investigation [13]
- Design solution strategy incorporating parallel analytical procedures
- Localize where the incident happened (affected manufacturing step)
- Determine how it happened (circumstances leading to incident)
- Establish why the incident occurred (underlying risks)
Preventive Measures [14]
- Implement corrective and preventive actions (CAPA) addressing root causes
- Establish monitoring systems to detect similar issues early
- Document lessons learned and update standard operating procedures
The Scientist's Toolkit: Essential Research Reagents and Equipment
The table below details key materials and equipment essential for continuous pharmaceutical manufacturing research and implementation:
| Item Name | Function/Purpose | Application Notes |
|---|---|---|
| Process Analytical Technology (PAT) | Enables real-time monitoring of critical quality attributes [6] | Essential for quality control in continuous processes; includes various sensors and analytical tools |
| Flow Reactor Systems | Provides uninterrupted flow of materials through integrated unit operations [11] | Superior for APIs with robust and scalable chemistry; may require customization for specific compounds |
| Scanning Electron Microscope with EDX | Identifies inorganic contaminants through surface topography and chemical analysis [13] | Critical for troubleshooting particle contamination; provides rapid, non-destructive analysis |
| LC-HRMS System | Separates and identifies complex organic compounds and degradation products [13] | Powerful for structure elucidation of contaminants; combines liquid chromatography with high-resolution mass spectrometry |
| Raman Spectroscopy | Non-destructive analysis of organic particles through database comparison [13] | Identifies organic contaminants quickly; requires reference materials for accurate comparison |
| Integrated Control Software | Manages and coordinates all unit operations in continuous system [11] | Enables real-time adjustments and data collection throughout manufacturing process |
| Powder Blending Systems | Creates homogenous mixture of API with excipients in continuous mode [6] | Reduces mixing time and improves blend homogeneity compared to batch systems |
The Expanding Role of PMI in Assessing Environmental Sustainability
Troubleshooting Guides
Inconsistent PMI Values
Problem: Significant variation in calculated PMI values for the same process across different experimental runs. Solution:
- Verify System Boundaries: Ensure all material inputs are consistently accounted for in both batch and continuous processes. Common omissions include water, solvents for cleaning-in-place (CIP), and buffers [2].
- Standardize Units: Confirm all mass inputs are in the same unit (e.g., kilograms) before summation. The denominator (mass of drug substance, DS) must also be in the same unit [2].
- Review Process Parameters: For continuous processes, ensure steady-state operation has been reached before data collection. Fluctuations in flow rates or concentrations during start-up/shutdown can skew results [16].
High PMI in Continuous Processes
Problem: A continuous process shows a higher PMI than its batch counterpart, contrary to expectations. Solution:
- Conduct Sensitivity Analysis: Assess the impact of individual parameters. A high solvent flow rate in flow chemistry is a common culprit that can worsen the environmental profile [16].
- Evaluate Productivity: PMI does not account for time. A continuous process with a higher PMI might produce more drug substance per unit time, making it more resource-efficient overall. Calculate the mass of DS produced per day or week for a more complete picture [2].
- Check for Over-Scaling: Laboratory-scale continuous equipment sometimes uses disproportionately large solvent volumes. Investigate miniaturization or solvent recycling strategies [16].
Incomplete Sustainability Assessment
Problem: Relying solely on PMI for environmental impact assessment. Solution:
- Integrate Energy Metrics: PMI does not capture energy consumption, a key driver of sustainability in biologics manufacturing. A batch process with a lower PMI may have a much higher energy footprint per gram of DS [2].
- Develop Complementary Metrics: Create a dashboard of metrics beyond PMI. This should include energy consumption (kWh/kg of DS), water consumption, and waste generation to provide a comprehensive view [2].
- Use a Holistic Framework: Apply Green Chemistry principles (e.g., the P5 Standard) to assess environmental, social, and economic impacts throughout the project lifecycle [17].
Frequently Asked Questions
What is Process Mass Intensity (PMI) and how is it calculated?
PMI is a key green chemistry metric that measures the efficiency of a manufacturing process. It is the total mass of materials used to produce a specified mass of a product. The formula is: PMI = Total Mass of Materials Entering Process (kg) / Mass of Product (kg) A lower PMI indicates a more efficient process with less waste. In pharmaceutical contexts, the product is typically the Drug Substance (DS) [2].
When comparing batch and continuous processes, is PMI sufficient?
No, PMI is a useful benchmarking tool but is not sufficient for a complete sustainability assessment. While it effectively measures material efficiency, it ignores other critical factors like energy consumption, process time, and cost [2]. A comprehensive evaluation should use PMI alongside other environmental and economic metrics. For instance, a continuous process might have a comparable PMI to a batch process but a much higher productivity rate, leading to a lower overall environmental impact per kilogram of drug produced over time [2].
Can a continuous process ever have a worse environmental profile than a batch process?
Yes. While continuous processing often offers sustainability advantages, it is not a guarantee. Research case studies have shown that increased solvent usage in flow mode can worsen the environmental profile compared to batch [16]. The outcome depends heavily on how the continuous process is designed and optimized. Therefore, it is crucial to evaluate each process on a case-by-case basis using robust metrics.
What are the best practices for presenting PMI and sustainability data?
- Use Clear Visuals: Employ bar charts for direct comparisons between batch and continuous PMI. Ensure high color contrast for accessibility [18].
- Provide Data Tables: Always supplement charts with data tables to ensure precise values are available and the information is accessible [18].
- Contextualize Findings: Explain what the PMI values mean in a broader context. For example, compare your results to industry benchmarks or highlight how a higher PMI might be offset by reduced energy use [2] [19].
Experimental Data and Protocols
Comparative PMI Analysis: Batch vs. Continuous
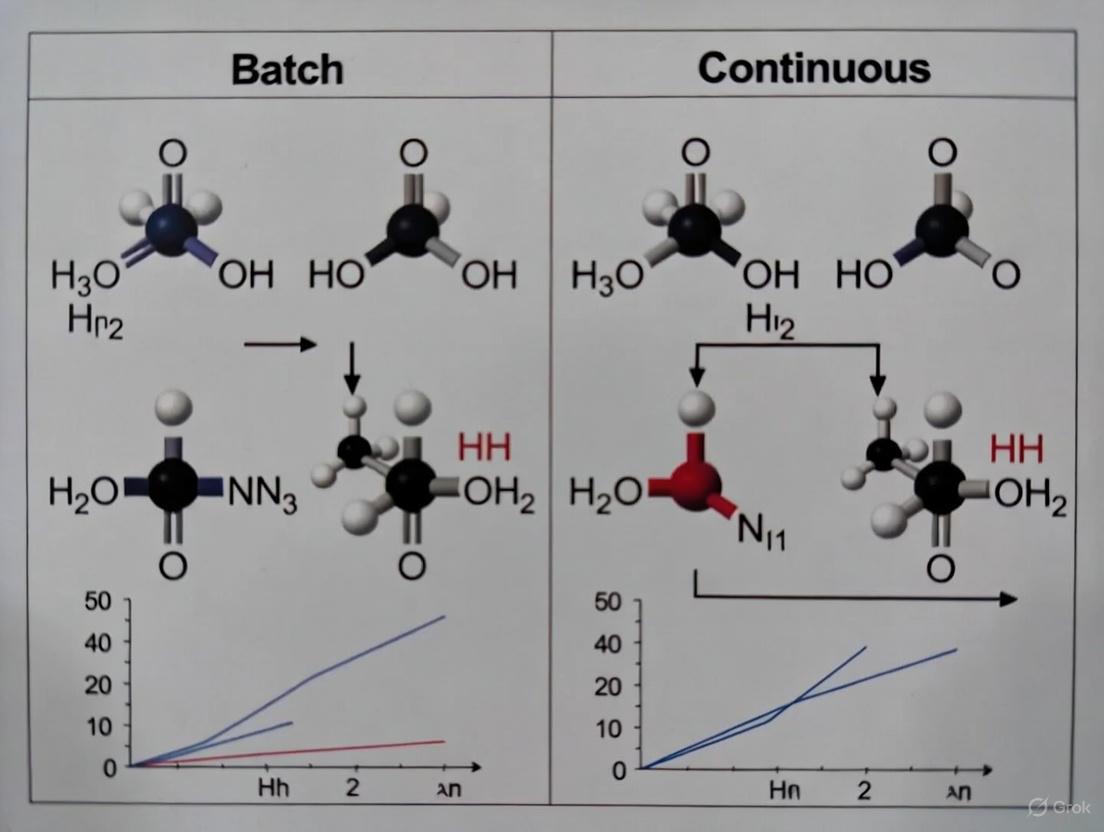
The table below summarizes example PMI values from research, illustrating that continuous processes can be comparable or superior to batch processes. These values are for illustration; actual PMI is highly process-dependent [2].
Table 1: Example PMI Values for mAb Production
| Process Type | Typical PMI Range (kg input / kg DS) | Key Influencing Factors |
|---|---|---|
| Batch | Comparable to Continuous | Bioreactor scale, cell culture titers, purification yield [2] |
| Continuous | Comparable to Batch | Perfusion rate, harvest cell density, resin capacity in continuous chromatography [2] |
| Intensified Batch | Lower than traditional Batch | Higher cell densities, process integration, reduced cycle time [2] |
Standard Protocol for Determining PMI in Biologics Processes
Objective: To calculate and compare the Process Mass Intensity (PMI) for batch and continuous manufacturing processes for monoclonal antibodies (mAbs).
Materials:
- Research Reagent Solutions:
- Cell Culture Media: Provides nutrients for cell growth and antibody production.
- Buffers & Salts: Maintain pH and ionic strength during purification steps.
- Chromatography Resins: Key for purifying the target antibody from the cell culture harvest.
- Cleaning Agents (CIP Solutions): Used to clean and sanitize equipment between batches or during continuous operation.
Methodology:
- Define Process Boundaries: Clearly state the start and end points of the analysis (e.g., from inoculum expansion to purified drug substance).
- Catalog Material Inputs: For a single batch or a defined period of continuous operation (e.g., 24 hours at steady state), record the mass of all inputs. This must include [2]:
- Water for injection (WFI)
- Cell culture media and feeds
- Buffers and solvents
- Chromatography resins and filters
- Cleaning and sanitization agents
- Measure Output: Accurately weigh or quantify the final mass of the purified drug substance (DS) produced.
- Calculate PMI: Use the formula PMI = Total Mass of Inputs / Mass of DS.
- Perform Sensitivity Analysis: Vary key process parameters (e.g., perfusion rate in continuous culture, buffer volumes in purification) to understand their impact on PMI [2] [16].
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for mAb Process Development
| Item | Function in Experiment |
|---|---|
| Chemically Defined Media | Supports consistent cell growth and protein production in bioreactors, reducing batch-to-batch variability. |
| Protein A Chromatography Resin | The core capture step in mAb purification; its binding capacity directly impacts the amount of resin needed and thus the PMI. |
| Ion Exchange Resins | Used for polishing steps to remove impurities like host cell proteins and DNA; a key determinant of final product purity. |
| Ultrafiltration/Diafiltration (UF/DF) Membranes | Used for buffer exchange and concentration of the final drug substance. |
| Process Analytical Technology (PAT) Tools | Probes and sensors for real-time monitoring of critical process parameters (e.g., pH, dissolved oxygen, metabolite levels), essential for controlling continuous processes. |
Process Visualization
PMI Assessment Workflow
Batch vs Continuous Flow
Methodologies for Calculating and Applying PMI in Process Design
Modeling and Simulation Tools for PMI Projection (e.g., PharmaPy)
Frequently Asked Questions (FAQs)
FAQ 1: What is PharmaPy and what is its primary role in PMI projection for pharmaceutical processes?
PharmaPy is an open-source, Python-based modeling platform specifically designed for the development of hybrid pharmaceutical manufacturing flowsheets [20] [21]. Its primary role in Process Mass Intensity (PMI) projection is to provide a versatile digital design tool that enables researchers and engineers to create mechanistic models of both batch and continuous drug substance manufacturing processes [20]. By simulating these processes, PharmaPy allows for the prediction of key performance indicators, including PMI, facilitating the comparison of resource efficiency between different manufacturing configurations (batch, continuous, or hybrid) before implementing them in a physical plant [20] [2].
FAQ 2: Can PharmaPy handle both batch and continuous process models for a direct PMI comparison?
Yes, this is a core capability of PharmaPy. It is designed with the flexibility to model and simulate purely batch, purely continuous, and hybrid manufacturing configurations within a single framework [20] [21]. This allows for a direct and consistent comparison of process economics and critical quality attributes, such as PMI, between different operational modes [20]. This capability is vital for research aimed at identifying the most efficient and sustainable manufacturing route based on quantitative data rather than prior beliefs about which mode is best [20] [2].
FAQ 3: What are the common numerical errors encountered during flowsheet simulation and how can they be resolved?
Common numerical errors often relate to the failure of the dynamic algebraic equation solvers. PharmaPy employs robust numerical integrators (SUNDIALS) through the Python package Assimulo for simulating ODEs and DAEs [21]. If simulations fail, users should:
- Check Model Consistency: Ensure the initial conditions and parameters are physically meaningful and consistent across all unit operations.
- Review Solver Tolerances: Adjust the relative and absolute tolerances for the ODE/DAE solver to balance between computational speed and solution accuracy.
- Simplify the Flowsheet: For hybrid processes, the interaction between discrete and continuous sections can cause discontinuities [20]. Simplifying the representation of certain unit operations or breaking down the simulation into smaller sections can help isolate and resolve the issue.
FAQ 4: How do I perform parameter estimation for my kinetic models within PharmaPy?
PharmaPy has an in-house implementation of the Levenberg-Marquardt algorithm for parameter estimation [21]. The methodology involves:
- Defining the Kinetic Object: Create a kinetic object that encapsulates the parameter values and expressions for the reaction or transformation mechanisms [20].
- Linking to Unit Operation: Aggregate this kinetic object with the relevant unit operation model (e.g., a reactor or crystallizer) [20].
- Providing Experimental Data: Supply the time-series experimental data against which the model parameters will be calibrated.
- Executing the Estimation: The software will then adjust the unknown parameters to minimize the difference between the model prediction and the experimental data.
FAQ 5: My optimization with an external solver is not converging. What should I check?
Since PharmaPy allows embedding simulations within external optimization packages, convergence issues can stem from several areas [20]:
- Simulation Robustness: Ensure the underlying PharmaPy flowsheet simulation is numerically stable and converges reliably for all potential input values the optimizer might try. A single simulation failure can cause the entire optimization to halt.
- Objective Function Formulation: Check that the objective function (e.g., minimizing PMI or maximizing yield) is correctly defined and scaled. Poorly scaled objectives can mislead the optimizer.
- Optimizer Choice: Confirm that the selected gradient-free optimizer is appropriate for the problem's characteristics (number of variables, presence of noise, etc.) [20]. It may be necessary to adjust the optimizer's settings or try a different algorithm.
Troubleshooting Guides
Issue 1: Flowsheet Simulation Failure in Hybrid Configurations
- Problem: The simulation fails or produces non-physical results when connecting a batch unit to a continuous unit, or vice-versa.
- Diagnosis: Hybrid processes inherently combine process dynamics and systemic mathematical discontinuities, which can challenge the sequential-modular solver [20].
- Solution: PharmaPy is designed to decouple continuous and discontinuous sections of a flowsheet [21]. Ensure that the material streams connecting different operational modes are correctly specified. Use the provided interpolation methods for handling the input and outputs between dynamic unit operation models to manage the transfer of material and information at the boundaries [20].
Issue 2: Inaccurate Parameter Estimation Results
- Problem: Estimated parameters from the Levenberg-Marquardt algorithm do not fit the experimental data well or show high uncertainty.
- Diagnosis: This can be caused by poor-quality data, an incorrectly specified kinetic model, or parameters that are insensitive (non-identifiable) given the available data.
- Solution:
- Verify Data Quality: Check experimental data for outliers and inconsistencies.
- Sensitivity Analysis: Perform a local sensitivity analysis to determine which parameters have the strongest influence on the model outputs. Focus estimation efforts on these.
- Model Re-specification: Review the kinetic expressions for physical consistency. The model structure itself may be incorrect.
Issue 3: Material Balance Errors in Sequential-Modal Simulation
- Problem: The overall material balance of the flowsheet does not close, indicating a loss or gain of mass.
- Diagnosis: In a sequential-modular approach, each unit is simulated individually in a pre-defined sequence, and errors can propagate [21].
- Solution:
- Unit-Checking: Isolate and simulate each unit operation independently to verify its individual material balance.
- Stream Specification: Double-check the definitions of all
StreamandPhaseobjects to ensure that flow rates, compositions, and densities are correctly passed from one unit to the next [20]. - Sequence Verification: Confirm that the sequence of unit operations in the flowsheet is logically correct.
Experimental Protocols & Data Presentation
Protocol 1: Methodology for Comparative PMI Analysis
Objective: To determine and compare the Process Mass Intensity (PMI) of a monoclonal antibody (mAb) process using batch and continuous manufacturing configurations.
Experimental/Modeling Workflow:
- System Definition: Define the overall process for mAb production, including key unit operations (e.g., bioreactor, purification steps).
- Flowsheet Configuration:
- Parameterization: Use consistent kinetic parameters (e.g., growth rates, reaction yields) for both configurations to ensure a fair comparison.
- Simulation Execution: Run dynamic simulations for the batch process and steady-state (or dynamic) simulations for the continuous process until a pre-defined amount of Drug Substance (DS) is produced.
- Data Collection: For each simulation, extract the total mass of raw materials input and the mass of the final DS produced.
- PMI Calculation: Calculate PMI for each configuration using the standard formula: PMI = Total Mass of Materials Input (kg) / Mass of Drug Substance (kg) [2].
The workflow for this comparative analysis is outlined in the following diagram:
Table 1: Sample PMI Comparison Data for mAb Processes
| Process Configuration | Total Mass Input (kg) | Mass of DS (kg) | PMI | Key Assumptions |
|---|---|---|---|---|
| Batch | 52,500 | 1.5 | 35,000 | 15,000 L bioreactor, 2 g/L titer, 50% yield |
| Continuous | 262,500 | 7.5 | 35,000 | 1,000 L bioreactor, 2 g/L titer, 50% yield |
| Continuous (Intensified) | 105,000 | 7.5 | 14,000 | 1,000 L bioreactor, 5 g/L titer, 70% yield |
Note: The data in Table 1 is illustrative. A key finding from research is that while base-case continuous processes may have a PMI comparable to batch, process intensification (e.g., higher titers, improved yields) can drive significant PMI improvements in continuous modes [2].
Protocol 2: Parameter Estimation for Crystallization Kinetics
Objective: To estimate the nucleation and growth kinetics of an Active Pharmaceutical Ingredient (API) from laboratory-scale experimental data.
Methodology:
- Experimental Data Collection: Conduct a series of lab-scale crystallization experiments, measuring crystal size distribution (CSD) and solute concentration over time under varying conditions (e.g., temperature, supersaturation).
- Unit Operation Setup: Instantiate a batch crystallizer unit model in PharmaPy.
- Kinetic Model Definition: Create a kinetic object with candidate expressions for nucleation (e.g., B = kb * ΔS^b) and growth (e.g., G = kg * ΔS^g) rates, where parameters (kb, b, kg, g) are to be estimated [20].
- Aggregation: Aggregate the kinetic object with the crystallizer unit model [20].
- Estimation Execution: Use the embedded Levenberg-Marquardt algorithm by providing the experimental data, allowing the software to find the parameter set that best fits the data [21].
The relationship between the unit model and kinetic model is shown below:
The Scientist's Toolkit: Research Reagent & Model Solutions
Table 2: Essential Components for a PharmaPy Flowsheet Model
| Item Name | Type | Function / Description |
|---|---|---|
| Phase Object | Software Class | Represents a material holdup within a piece of equipment, defining its state and composition [20]. |
| Stream Object | Software Class | Represents flowing material connecting unit operations, carrying state information between them [20]. |
| Unit Operation (UO) Object | Software Class | A model of a specific processing step (e.g., reactor, crystallizer). It can aggregate Material and Kinetic objects [20]. |
| Kinetic Object | Software Class | Encapsulates parameter values and expressions for kinetic or transport mechanisms (e.g., reaction rates, crystal growth) [20]. |
| SUNDIALS/Assimulo | Numerical Library | Provides robust ODE/DAE numerical integrators for simulating the dynamic behavior of unit models [21]. |
| Levenberg-Marquardt Algorithm | Tool | In-house parameter estimation algorithm for calibrating model parameters to experimental data [21]. |
Establishing Key Critical Quality Attributes (CQAs) for Analysis
Defining Critical Quality Attributes (CQAs)
A Critical Quality Attribute (CQA) is a physical, chemical, biological, or microbiological property or characteristic that must be maintained within an appropriate limit, range, or distribution to ensure the desired product quality [23] [24]. CQAs are fundamental to the FDA's Process Analytical Technology (PAT) framework and the Quality by Design (QbD) approach, which emphasize building quality into a product during its design and manufacturing processes, rather than relying solely on final product testing [23] [25].
In the context of comparing batch versus continuous manufacturing processes, particularly in research measuring Process Mass Intensity (PMI), establishing and monitoring CQAs is essential. It ensures that process changes aimed at improving sustainability (lower PMI) do not adversely affect the critical quality of the resulting biologic or drug substance [2] [26] [27].
Categories and Examples of CQAs
The following table outlines common categories and specific examples of CQAs for biopharmaceuticals, which are critical for both batch and continuous processes.
| Category | Description | Specific CQA Examples |
|---|---|---|
| Product-Related Variants [23] [24] | Molecular characteristics of the drug product itself. | Size variants, charge variants, glycan patterns, oxidation levels [23]. |
| Safety-Related (Purity & Impurities) [23] [24] | Measures of unwanted process-related materials that impact product safety. | Host Cell Proteins (HCP), DNA, leachables, endotoxins [23] [24]. |
| Identity & Potency [24] | Confirmation of the correct product and its biological activity. | Product titer, composition, strength, and biological activity measured by potency assays [24]. |
| Sterility [24] | Freedom from viable contaminating microorganisms. | Bioburden, mycoplasma, absence of adventitious agents [24]. |
Frequently Asked Questions (FAQs) and Troubleshooting
1. How do we define CQAs for a new product in development? CQAs are identified through a risk-based approach that begins in early development. Initially, limits may be broader and are refined as more process and product understanding is gained [24] [25]. The process involves:
- Identifying Quality Attributes: List all potential physical, chemical, biological, and microbiological properties.
- Risk Assessment: Evaluate the impact of each attribute on safety and efficacy. Attributes with a high potential impact are designated as "critical."
- Linking to Process: Understand how Critical Process Parameters (CPPs) and Critical Material Attributes (CMAs) influence the CQAs [27].
2. Our continuous process shows a favorable PMI, but we are observing higher variability in a charge variant CQA. What could be the cause? In continuous manufacturing, Process Analytical Technology (PAT) tools are crucial for real-time monitoring and control [23] [27]. Higher variability may indicate:
- Inadequate PAT: The in-line or at-line sensors monitoring the CQA may not be providing sufficiently timely data for the control system to respond.
- Improper Control Strategy: The feedback loop between the PAT tool and the process control parameters may need optimization to correct deviations more rapidly.
- Residence Time Distribution (RTD): Understanding the RTD—the distribution of time materials spend in the system—is critical in continuous processes to trace and control the source of variability [27].
3. For a continuous process, is quality control performed differently than in a batch process? Yes, the paradigm shifts significantly.
- Batch Process: Quality control is typically performed at the end of a production run on samples from the completed batch [28] [29] [4].
- Continuous Process: Quality control is integrated and ongoing. PAT tools are used for real-time monitoring of CQAs, allowing for immediate detection and correction of quality issues without stopping production [28] [27]. This aligns with the real-time release testing (RTRT) concept.
4. We are transitioning from batch to a hybrid process. Can we use the same CQAs? Yes, the fundamental CQAs for a product (e.g., purity, potency) remain the same regardless of the manufacturing mode [27]. However, your control strategy and the methods for monitoring them will likely need to evolve. You may need to:
- Implement new PAT tools for real-time measurement.
- Establish new control limits for CPPs that are specific to the continuous or hybrid unit operations.
- Develop a deep understanding of how material attributes (CMAs) like powder flowability impact CQAs in a continuous feed system [27].
CQA Control Strategy in Manufacturing
The following diagram illustrates the logical relationship and feedback loop between process parameters, material attributes, and quality attributes in a controlled manufacturing process, which is central to both batch and continuous modes.
Experimental Protocol: Linking Process Parameters to a Purity CQA
This protocol outlines a general methodology for characterizing how a Critical Process Parameter (CPP) affects a purity-related CQA (e.g., Host Cell Protein level).
1. Objective: To determine the impact and establish a control range for a critical process parameter on a key CQA.
2. Materials and Reagents:
| Item | Function |
|---|---|
| Chromatography System (e.g., AKTA) | For purifying the drug substance from process impurities. |
| ELISA Kit for HCP | A specific assay to quantify levels of host cell proteins, a common purity CQA [24]. |
| Cell Culture Samples | In-process samples containing the product and impurities. |
| Buffers and Eluents | Mobile phases for the chromatography process. |
3. Methodology:
- Design of Experiments (DoE): Use a structured DoE approach instead of a one-factor-at-a-time analysis. For example, a Full Factorial Design can be used to study parameters like pH and conductivity of the elution buffer.
- Process Execution: Perform multiple small-scale purification runs using the chromatography system. The input CPPs (pH, conductivity) are varied according to the DoE matrix.
- Sample Collection: For each run, collect the elution fraction containing the drug substance.
- CQA Analysis: Analyze each elution fraction using the HCP ELISA kit to determine the level of this impurity, which is the measured output for the CQA.
- Data Analysis: Statistically analyze the results (e.g., using ANOVA and regression modeling) to build a model that predicts the HCP level (CQA) as a function of the input CPPs. This model will define the proven acceptable range for the CPPs to ensure the CQA (HCP level) stays within its specified limit.
Research Reagent Solutions for CQA Analysis
The following table details key reagents and assays used in the experimental characterization of CQAs.
| Research Reagent / Assay | Function / Brief Explanation |
|---|---|
| Host Cell Protein (HCP) ELISA | Quantifies residual process-related impurities to ensure product purity and safety [24]. |
| Potency Assay (e.g., cell-based bioassay) | Measures the biological activity of the drug product, a direct indicator of its potency [24]. |
| Charge Variant Analysis Kit (e.g., iCIEF) | Characterizes the distribution of charge variants (e.g., deamidation) which can impact product stability and efficacy [23]. |
| Glycan Analysis Reagents | Used to characterize the glycosylation pattern of a biologic, a CQA that can affect safety (immunogenicity) and efficacy [23]. |
| Endotoxin Testing Kit (LAL) | Detects and quantifies bacterial endotoxins, a critical safety-related CQA for parenteral products [24]. |
Technoeconomic Analysis (TEA) Frameworks Integrating PMI
Frequently Asked Questions (FAQs)
1. What is the primary purpose of integrating Process Mass Intensity (PMI) into a Techno-Economic Analysis (TEA) framework? Integrating PMI into TEA provides a more holistic view of a process's viability. While TEA assesses economic feasibility through capital and operating costs, PMI measures material efficiency and environmental impact by calculating the total mass of materials used per unit of product. Their integration is crucial for sustainable process design, allowing researchers to understand the trade-offs between economic performance and environmental footprint, particularly when comparing traditional batch and emerging continuous manufacturing processes [30] [31].
2. In the context of batch vs. continuous pharmaceutical manufacturing, what are the key economic and environmental advantages of continuous processing? Continuous manufacturing often demonstrates superior economic and environmental performance. Techno-economically, it can reduce production time by 70-90%, lower variable costs by 40-50%, and improve energy efficiency by up to 97% for some APIs like ibuprofen [32] [31] [11]. From a PMI and environmental perspective, it leads to significant waste reduction, lowers water consumption by 25-50%, and can reduce carbon emissions due to more efficient energy and material use [32] [31].
3. What is a major data quality challenge when performing TEA at low Technology Readiness Levels (TRLs), and how can it be mitigated? At low TRLs, a major challenge is data uncertainty and scarcity. Processes, especially emerging ones like continuous manufacturing, are often defined by laboratory-scale experiments, making it difficult to project accurate capital and operating costs or precise material balances for PMI calculation [30]. This can be mitigated by using surrogate modeling and structured uncertainty analysis. Building surrogate models based on limited experimental data can help predict key performance indicators, while techniques like Monte Carlo simulation can be employed to understand how uncertainty propagates through the TEA model, providing a range of possible economic outcomes instead of a single, potentially misleading, figure [33].
4. My TEA model shows that a continuous process has higher capital costs than batch. Does this mean it is not economically viable? Not necessarily. A higher capital expenditure (CAPEX) for continuous manufacturing must be evaluated against its operational benefits [31] [11]. Continuous processes often have significantly lower operating expenditures (OPEX) due to reduced labor, higher energy efficiency, and lower waste handling costs. A full TEA will calculate metrics like Net Present Value (NPV) or Internal Rate of Return (IRR) over the project's lifetime. The substantial OPEX savings of a continuous process can often justify the higher initial investment. Furthermore, the integrated TEA-PMI framework might reveal additional value from sustainability benefits, such as a lower environmental burden, which can be a strategic advantage [31].
5. How can I define a consistent functional unit for an integrated TEA and PMI comparison between batch and continuous processes? The functional unit is critical for a fair comparison. For pharmaceutical manufacturing, the most appropriate functional unit is typically per mass unit of final, purified Active Pharmaceutical Ingredient (API) (e.g., per kilogram of 99.9% pure ibuprofen) [31]. This ensures that all material inputs (for PMI calculation) and energy/utility costs (for TEA) are normalized to an equal output basis. It is crucial that the quality and purity of the API are identical for both the batch and continuous processes being compared to ensure the analysis is valid [31].
Troubleshooting Guides
Issue 1: Inconsistent or Incomparable Results in Batch vs. Continuous TEA
Problem: The TEA results for batch and continuous processes are inconsistent, making a fair comparison impossible. This often stems from differing system boundaries or assumptions.
Solution:
- Step 1: Harmonize System Boundaries. Ensure both analyses use a "cradle-to-gate" approach, starting from the same raw material inputs and ending with the same final product (e.g., finished tablet or purified API). All major unit operations must be included for both processes [30] [31].
- Step 2: Standardize the Functional Unit. Re-base all calculations on a consistent functional unit, such as "per kilogram of final product" [31].
- Step 3: Align Financial Assumptions. Verify that key financial parameters are identical:
- Plant lifetime (e.g., 20 years)
- Annual operating hours
- Discount rate for NPV calculations
- Cost basis for utilities, labor, and raw materials (from the same year and source) [30]
Issue 2: Difficulty in Accurately Estimating PMI for a New Continuous Process
Problem: For a novel continuous process at a low TRL, there is insufficient data to calculate a reliable PMI, as the process is not yet optimized.
Solution:
- Step 1: Develop a Detailed Process Model. Use simulation software (e.g., Aspen Plus) to create a mass and energy balance based on experimental data, even if limited. This model will identify all material inputs and outputs [31].
- Step 2: Calculate a Theoretical PMI. Use the output of the process model to calculate the total mass of inputs (raw materials, solvents, catalysts) divided by the mass of the product. Clearly document this as an initial, non-optimized PMI [31].
- Step 3: Perform Sensitivity Analysis. Identify which materials contribute most to the PMI. This highlights the "hot spots" where process optimization—such as solvent recycling or catalyst recovery—will have the greatest impact on reducing PMI and operating costs, guiding future R&D efforts [33].
Issue 3: High Capital Cost (CAPEX) for Continuous Manufacturing Equipment
Problem: The initial investment for continuous flow reactors and integrated monitoring systems is prohibitively high, making the TEA results unfavorable.
Solution:
- Step 1: Explore Modular and Open-Source Platforms. Investigate the use of modular continuous systems that can be scaled out rather than up, potentially reducing initial capital outlay. Open-source platforms like BioProcessNexus can also provide lower-cost modeling and analysis tools [33].
- Step 2: Quantify OPEX Savings and Intangible Benefits. Rigorously model the operational savings from continuous processing, which often offset high CAPEX. Key areas to calculate include:
- Step 3: Justify via Lifecycle Costing. Present the TEA results using lifecycle costing (Total Cost of Ownership) over a 10-20 year period, which will capture the long-term OPEX benefits that make the continuous process more economical overall [30] [11].
Experimental Protocols & Data
Quantitative Comparison: Batch vs. Continuous Manufacturing
The following table summarizes key quantitative findings from recent studies comparing batch and continuous pharmaceutical manufacturing, which are essential for populating TEA and PMI models [32] [31] [11].
| Metric | Batch Performance | Continuous Manufacturing Performance | Improvement |
|---|---|---|---|
| Production Time | Reference (Weeks/Months) | 1 Day | Reduction of 70-90% [32] |
| Production Cost (Variable) | Reference | Reduction of 40-50% [11] | |
| Energy Consumption | Reference (e.g., for Ibuprofen) | Reduction of up to 97% [31] | |
| Facility Space | Reference | Reduction of 30-50% [32] | |
| Water Consumption | Reference | Reduction of 25-50% [32] | |
| Dose Uniformity | Reference | Improvement of ~40% [32] |
Detailed Methodology for Comparative Dissolution Testing
This protocol is critical for assessing a Critical Quality Attribute (CQA) when comparing tablets made via batch and continuous direct compression, which can impact bioperformance and process validation [34].
- 1. Objective: To compare the dissolution profiles of immediate-release tablets manufactured by Batch Direct Compression and Continuous Direct Compression (CDC) using identical formulations.
- 2. Materials:
- API (e.g., Ibuprofen 50, a BCS Class II drug)
- Fillers: Microcrystalline Cellulose (MCC) and Dibasic Calcium Phosphate (DCP) or Lactose Monohydrate
- Disintegrant: Croscarmellose Sodium
- Glidant: Colloidal Silica
- Lubricant: Sodium Stearyl Fumarate
- 3. Equipment:
- Batch Blender (e.g., Turbula mixer) and Batch Press
- Continuous Direct Compression Line (e.g., with feeders, continuous blender, tablet press)
- USP-Apparatus 2 (Paddles)
- UV-Vis Spectrophotometer or suitable HPLC system
- 4. Experimental Design (DoE):
- Factors: Use a Design of Experiments approach. Key factors should include:
- Manufacturing Mode (Batch vs. CDC)
- Blender Speed (e.g., 200 rpm vs. 400 rpm)
- Tablet Tensile Strength (e.g., 2 MPa vs. 2.5 MPa)
- Disintegrant Concentration
- Filler Combination (MCC/DCP vs. MCC/Lactose)
- Response Variable: Dissolution profile (e.g., % API released at 5, 10, 15, 20, 30, and 45 minutes).
- Factors: Use a Design of Experiments approach. Key factors should include:
- 5. Procedure:
- Blending: For each run in the DoE, prepare the powder blend using the specified method (batch or continuous) and settings.
- Compression: Compress tablets to the target tensile strength on the respective press.
- Dissolution Testing: Perform dissolution testing on the tablets in 900 mL of phosphate buffer (pH 7.2) at 37°C, with a paddle speed of 50 rpm. Sample at predetermined time points.
- Analysis: Quantify the amount of API released at each time point using the analytical method.
- 6. Data Analysis:
- Plot and compare mean dissolution profiles.
- Use a model-independent method (e.g., similarity factor f2) to statistically compare profiles. An f2 value greater than 50 suggests similar dissolution behavior.
- Use multivariate analysis (e.g., Partial Least Squares regression) to determine the relative impact of the manufacturing mode versus other formulation and process variables on the dissolution behavior [34].
Frameworks and Workflows
TEA-PMI Integration Workflow
The following diagram illustrates the iterative workflow for integrating TEA and PMI analysis, which is especially valuable for evaluating and optimizing new processes like continuous manufacturing.
Experimental Design for Comparative Studies
This diagram outlines the logical flow for designing an experiment that generates data suitable for a robust batch vs. continuous TEA and PMI comparison.
The Scientist's Toolkit: Key Research Reagent Solutions
The table below lists essential materials and their functions for conducting experiments related to solid dosage form manufacturing, which is a common context for batch vs. continuous comparison studies [34] [31].
| Item | Function in Research | Critical Consideration for TEA/PMI |
|---|---|---|
| Active Pharmaceutical Ingredient (API) (e.g., Ibuprofen) | The biologically active component of the drug product. | A high-cost, low-availability API favors continuous manufacturing development to save material during R&D [34]. |
| Excipients (e.g., MCC, Lactose, DCP) | Inactive ingredients that formulate the API into a functional dosage form. | Powder flow properties (e.g., of MCC) are critical for continuous processing reliability and content uniformity [34]. |
| Disintegrant (e.g., Croscarmellose Sodium) | Promotes the breakup of a tablet in the gastrointestinal tract. | Its concentration is a key variable in DoE; impacts dissolution performance in both batch and continuous modes [34]. |
| Lubricant (e.g., Sodium Stearyl Fumarate) | Reduces friction during tablet compression and ejection. | Over-lubrication can negatively affect tablet hardness and dissolution; optimal level is process-dependent [34]. |
| Organic Solvents (e.g., for API synthesis) | Used in chemical reactions and purification steps. | A major contributor to PMI. Opportunities for recycling in continuous processes can drastically reduce PMI and OPEX [31]. |
What is PMI and why is it a critical metric in pharmaceutical crystallization? Process Mass Intensity (PMI) is a key Green Chemistry metric that measures the total mass of materials (solvents, reagents, etc.) used to produce a unit mass of the final product. In API crystallization, a lower PMI indicates a more efficient and environmentally sustainable process, as it signifies less waste generation and better resource utilization. PMI has become an indispensable tool for comparing the environmental and economic performance of different manufacturing approaches, particularly in the ongoing evaluation of batch versus continuous crystallization technologies [35] [36].
Troubleshooting Guides and FAQs
FAQ 1: Why is controlling polymorphism so critical in API crystallization like paracetamol, and how does it relate to PMI?
Polymorphism directly impacts both product quality and process efficiency. Different polymorphs can have vastly different physicochemical properties.
- Impact on Manufacturing: For paracetamol, Form I has poor compression properties, requiring extra binders during tablet formulation, whereas Form II can be directly compressed [37]. Producing the wrong polymorph can lead to batch rejection, reworking, and increased PMI due to wasted materials and extra processing steps.
- Process Stability: Form II paracetamol is metastable and can transform to the stable Form I, especially when contaminated with Form I, exposed to high moisture, or undergoing mechanical grinding [38]. This instability can lead to failed batches and increased material usage to meet quality specs, negatively affecting PMI.
FAQ 2: We are experiencing low product yield in our crystallization. What are the main causes and how can we improve it?
Low yield directly increases PMI by wasting valuable API. Common causes and solutions include:
- Excessive Solvent: Using too much solvent leaves a significant amount of compound dissolved in the mother liquor [39].
- Solution: Boil off a portion of the solvent and cool the solution again to perform a "second crop" crystallization. Ensure you are not using an excessive amount of solvent to dissolve semi-insoluble impurities during hot filtration [39].
- Rapid Crystallization: If crystallization is too quick, it can trap impurities within the crystal lattice, potentially leading to impure product and low effective yield after purification [39].
- Solution: Slow down crystallization by using a slight excess of hot solvent, transferring the solution to an appropriately sized flask to create a deeper solvent pool, and insulating the flask during cooling [39].
FAQ 3: Our crystallization process is inconsistent and lacks reproducibility. What factors should we control?
Inconsistency is a major driver of high PMI due to batch failures and re-processing.
- Feed Quality: Monitor and control the composition, concentration, pH, temperature, and dissolved solids of the feed stream to the crystallizer, as it is the primary source of impurities [40].
- Supersaturation Control: Operate within the metastable zone by controlling the cooling rate. Allowing the solution to enter the unstable zone leads to spontaneous, rapid crystallization that is difficult to control [41]. Using real-time refractive index monitoring can help track supersaturation and identify the ideal seeding point [41].
- Seeding: The use of seeds, especially for metastable forms like paracetamol Form II, is a common strategy to ensure consistent polymorphic outcome. Seeding should be done at the correct supersaturation level with the appropriate seed mass and crystal size [38] [36].
Quantitative PMI Comparison: Batch vs. Continuous Crystallization
Technoeconomic studies directly compare the performance of batch and continuous crystallization for APIs like paracetamol. The table below summarizes key findings from such analyses.
Table 1: Technoeconomic and Environmental Comparison of Batch and Continuous Crystallization for Paracetamol
| Metric | Batch Crystallization | Continuous Crystallization (MSMPR/COBC) | Notes & Sources |
|---|---|---|---|
| Process Mass Intensity (PMI) | Generally lower PMI [35] | Can have higher PMI, but shows better potential for expansion and optimization [35] | PMI is used as a quantified metric of environmental impact [35]. |
| Capital Expenditure (CapEx) | Lower overall cost for standard production volumes [35] | Cost is highly sensitive to antisolvent use rate and seed mass loading [36] | Continuous systems may offer cost benefits when scaling up [35]. |
| Operational Expenditure (OpEx) | Continuous methods can achieve higher yields and purities, decreasing processing times [36] | ||
| Process Performance & Scalability | Mature technology, but can have batch-to-batch variability [38] | Improved heat/mass transfer, better reproducibility, and smaller equipment size [38] [36] | Continuous Oscillatory Baffled Crystallisers (COBCs) offer improved performance over plug flow and batch units [36]. |
Detailed Experimental Protocols
Protocol 1: Seeded Cooling Crystallization of Paracetamol Form II using Additives
This protocol is adapted from research on the selective polymorphic formation of paracetamol Form II with the assistance of carboxylic acid additives [38].
Objective: To reproducibly produce the metastable, but more compressible, Form II of paracetamol on a large scale. Principle: Additives like fumaric acid and oxalic acid can assist in the selective nucleation and stabilization of Form II by forming solution complexes and extending the range of supersaturation for its nucleation [38].
Materials and Equipment:
- Paracetamol (API)
- Fumaric acid (additive)
- Aqueous solvent (water)
- Heated reactor with agitation and temperature control
- Seeding crystals of paracetamol Form II
Procedure:
- Solution Preparation: Prepare a solution of paracetamol in a 50 wt% aqueous fumaric acid solution with a paracetamol concentration of 44 mg/mL [38].
- Saturation: Heat the solution to ensure complete dissolution of all solids.
- Cooling and Seeding: Cool the solution to the predetermined seeding temperature. Introduce a seed crystal of pure paracetamol Form II to initiate controlled crystallization.
- Crystal Growth: Continue a controlled cooling profile to promote crystal growth while maintaining the supersaturation within the metastable zone for Form II.
- Isolation: Once crystallization is complete, isolate the crystals by filtration. The presence of fumaric acid in the mother liquor requires subsequent purification steps to ensure the chemical purity of the paracetamol product [38].
Protocol 2: Workflow for Solvent Screening and Solubility Measurement
A systematic workflow accelerates robust crystallization process development by minimizing material usage and development time [42].
Objective: To efficiently identify optimal solvents and antisolvents for a crystallization process that delivers high purity, yield, and low PMI. Principle: Combines in-silico predictions with high-throughput experimentation to build a accurate, system-specific solubility model [42].
The following diagram illustrates the sequential, iterative workflow for solvent screening and solubility measurement:
Materials and Equipment:
- API (e.g., Paracetamol, used for workflow validation [42])
- Candidate solvents and antisolvents
- High-throughput screening platform (e.g., with computer vision for automated solubility assessment [42])
- Analytical tools (e.g., HPLC for concentration measurement)
Procedure:
- In-Silico Prediction: Use structure-based software tools to predict API solubility in a wide range of potential solvents and generate a shortlist of feasible candidates [42].
- Initial Experimental Measurement: Measure the API solubility in the shortlisted solvents at room temperature using a high-throughput platform. This data is used to refine the initial predictions [42].
- Condition Selection: Use the refined solubility data to generate candidate solubility curves and determine potential crystallization conditions (e.g., solvent-antisolvent pairs, temperature ranges, expected yields) [42].
- Detailed Solubility Modeling: Select the most promising option and perform detailed solubility measurements across a range of temperatures. Use regression to build a precise, system-specific solubility model [42].
- Process Definition: Use the final model to define the optimal crystallization process parameters, such as the seeding point and cooling profile, for implementation and control [42].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagents and Materials for API Crystallization Research and Development
| Item | Function / Purpose | Example in Context |
|---|---|---|
| Carboxylic Acid Additives | Assist in selective polymorphic nucleation of metastable forms. | Fumaric acid and oxalic acid help nucleate and stabilize paracetamol Form II [38]. |
| Seed Crystals | Provide a controlled surface for crystal growth, ensuring consistent polymorphic form and particle size distribution. | Seeding a paracetamol solution with Form II crystals is crucial for reproducible Form II production [38] [36]. |
| Process Analytical Technology (PAT) | Enables real-time monitoring and control of Critical Process Parameters (CPPs) to ensure Critical Quality Attributes (CQAs). | Refractive Index sensors monitor supersaturation in real-time for optimal seeding and crystal growth control [41]. |
| Antisolvents | A substance added to reduce the solubility of the API, inducing crystallization. | The type and quantity of antisolvent are key variables optimized in continuous crystallizer design [36]. |
| Modeling & Simulation Software | Used for technoeconomic analysis, process optimization, and predicting outcomes like PMI and CapEx. | Python-based tools (e.g., PharmaPy) are used for simulation-optimization of crystallization flowsheets [35]. |
Frequently Asked Questions (FAQs)
FAQ 1: What is the fundamental difference between batch and continuous processing during scale-up?
Batch processing involves producing a specific quantity of material in a single, discrete run with a defined start and end point. Quality control is typically performed at the end of each batch, and equipment can be reconfigured between batches for different products [4] [28]. In contrast, continuous processing involves an ongoing, uninterrupted production flow where raw materials are constantly fed into the system and finished product continuously emerges. It relies on real-time monitoring and automated controls to maintain quality throughout the operation [6] [43].
FAQ 2: When should I consider continuous manufacturing over batch processing for my drug product?
Continuous manufacturing is advantageous for high-volume production with stable demand, where consistency, efficiency, and lower production costs are critical [4] [44]. It is particularly suitable for oral solid dosage (OSD) forms and can reduce quality control time by 50-70% and power consumption by up to 40% [6]. However, it requires significant initial investment and is less flexible for product changes. Batch processing remains ideal for small-to-medium volumes, products requiring high customization, or markets where flexibility and quality control for each discrete unit are paramount [4] [28].
FAQ 3: What are the most critical parameters to monitor when scaling up a milling process for active pharmaceutical ingredients (APIs)?
The most critical parameter is often the Particle Size Distribution (PSD), as it can directly impact bioavailability and dissolution rates [45]. During scale-up, factors like shear forces, heat transfer, and flow patterns change with larger volumes, risking dead zones or inconsistent mixing [46]. Using equipment certified for scalable performance, with consistent power, speed, and size characteristics from lab to production, is essential to maintain PSD and other critical quality attributes (CQAs) [45].
Troubleshooting Guides
Problem 1: Inconsistent Product Quality After Scale-Up
- Symptoms: Variation in critical quality attributes (CQAs) like viscosity, texture, or potency between laboratory batches and larger pilot/production batches.
- Possible Causes:
- Solutions:
- Pilot Trials: Conduct pilot-scale trials to fine-tune process parameters like mixing speed, time, and temperature [46].
- Equipment Assessment: Ensure the industrial mixer type (e.g., High Shear, Planetary) is appropriate for the product's viscosity and rheology [46].
- Process Analytical Technology (PAT): Implement PAT tools for real-time monitoring and control of CQAs during the run [6].
Problem 2: Failure to Maintain a "State of Control" and Meet Regulatory Standards
- Symptoms: Difficulties during regulatory inspections, failure to demonstrate process validation, or inability to maintain the "validated state" of equipment and systems.
- Possible Causes:
- Inadequate Documentation: Lack of a comprehensive Validation Master Plan (VMP) or failure to keep it as a "living document." [47]
- Poor Change Control: No formalized system for managing, testing, and documenting changes to equipment or processes [47].
- Insufficient Qualification: Equipment not properly qualified through Installation, Operational, and Performance Qualification protocols [47].
- Solutions:
- Develop a VMP: Create a detailed Validation Master Plan that acts as a project plan for all validation activities, defining scope, responsibilities, and milestones [47].
- Follow the "V-Diagram": Ensure specifications are precisely linked to qualification protocols [47].
- Implement Robust SOPs: Develop clear Standard Operating Procedures and train all team members to ensure compliance [46].
Problem 3: Managing Heat and Mass Transfer Inefficiencies at Larger Scales
- Symptoms: Inconsistent reaction outcomes, localized overheating, or extended processing times not seen at the lab scale.
- Possible Causes:
- Surface-to-Volume Ratio: This ratio decreases with scale, making heat removal less efficient [48].
- Improper Equipment Sizing: Reactors or mixing vessels are not designed for optimal heat exchange or mass transfer at the target production volume.
- Solutions:
- Advanced Engineering: Design and select equipment with efficient heating/cooling systems (e.g., jacketed reactors) and impellers optimized for mass transfer [48].
- Process Optimization: Use a step-wise scale-up approach (e.g., 10x increments per SUPAC guidelines) to carefully adjust parameters instead of a single, large jump [45] [48].
Quantitative Data Comparison: Batch vs. Continuous
The following table summarizes key quantitative differences between batch and continuous processes, which is critical for informing Process Mass Intensity (PMI) and sustainability assessments.
| Parameter | Batch Process | Continuous Process | Source |
|---|---|---|---|
| Typical Production Volume | Small to medium volumes [4] | Large-scale, high-volume output [4] | [4] |
| Production Rate | Slower, with stops and starts between batches [28] | Higher throughput, shorter processing times [4] | [4] [28] |
| Quality Control Time | N/A | Can be reduced by 50-70% [6] | [6] |
| Power Consumption | N/A | Can be reduced by up to 40% [6] | [6] |
| Process Mass Intensity (PMI) for mAbs | Comparable to continuous processes [2] | Comparable to batch processes [2] | [2] |
| Initial Capital Cost (CAPEX) | Lower initial setup cost [4] | Significant initial investment required [4] [44] | [4] [44] |
| Unit Cost | Higher unit costs [28] | Lower unit costs due to higher production rates [4] [28] | [4] [28] |
| Regulatory Approvals (as of 2022) | Traditional, well-established pathway | Only 7 drugs approved via CM by major agencies (FDA, EMA) [6] | [6] |
Experimental Protocols for Key Scale-Up Experiments
Protocol 1: Direct Compression Process Comparison for Oral Solid Dosage Forms
This protocol is used to compare the blend homogeneity and API uniformity of a formulation processed via batch and continuous direct compression.
- Objective: To compare the critical quality attributes of tablets produced by batch and continuous direct compression processes.
- Materials:
- Active Pharmaceutical Ingredient (API)
- Excipients (e.g., filler, disintegrant, lubricant)
- Batch Blender (e.g., "tumbling" V-blender)
- Continuous Powder Blending Line (e.g., with gravimetric feeders and continuous mixer)
- Tablet Press
- Methodology:
- Formulation: Use identical ratios of API and excipients for both processes.
- Batch Process:
- Load all powders into the batch blender.
- Blend for a predefined time at a fixed speed.
- Discharge the blend and compress into tablets.
- Continuous Process:
- Calibrate gravimetric feeders to ensure a continuous, steady flow of each ingredient.
- Pass the powder stream through the continuous mixer.
- Direct the output blend continuously to the tablet press for compression.
- Analysis: Use an UV-Vis reflectance method with chemometrics to analyze blend homogeneity and API content uniformity in the final tablets [6]. Particle size distribution of the final blend should also be measured.
- Key Parameters to Record:
- Mixing/Residence Time
- Blender RPM/Speed
- Resulting Blend Homogeneity
- API Uniformity in Final Tablet
Protocol 2: Pilot-Scale Mixing and Dynamics Validation
This protocol is essential for identifying and mitigating scaling risks related to mixing before full-scale production.
- Objective: To validate mixing parameters and product consistency on a pilot-scale machine before committing to a full production line.
- Materials:
- Pilot-scale mixer (e.g., Vacuum Emulsifying Mixer, Planetary Mixer) that is geometrically similar to the production-scale equipment.
- All raw materials for the product.
- In-process analytical tools (e.g., viscometer, pH meter, PAT probes).
- Methodology:
- Baseline Establishment: Document all critical parameters from the lab-scale process (mixing speed, time, temperature, shear rate, sequence of addition).
- Pilot Trials:
- Run multiple batches on the pilot-scale equipment.
- Systematically vary one parameter at a time (e.g., mixing speed, time) to find the optimal operating range.
- Sample the product at different stages and locations in the mixer to check for uniformity.
- Quality Assessment: Test all pilot batches against the target CQAs (viscosity, stability, texture).
- Key Parameters to Record:
- Power consumption of the mixer.
- Temperature profile throughout the batch.
- Final product viscosity/pH/texture.
- Presence of air bubbles or inhomogeneities.
Process Visualization and Workflows
Scale-Up Methodology Workflow
Batch vs. Continuous Decision Logic
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item / Solution | Function in Scale-Up |
|---|---|
| Vacuum Emulsifying Mixers | Used for creating stable creams, ointments, and emulsions; removes air bubbles to ensure product consistency and stability during scale-up [46]. |
| Planetary Mixers | Ideal for mixing high-viscosity or thick materials (e.g., pastes, doughs) that are difficult to homogenize with standard stirrers [46]. |
| Homogenizers | Provide high-shear mixing for particle size reduction and creating stable emulsions, critical for ensuring batch-to-batch uniformity [46]. |
| Process Analytical Technology (PAT) | A system for real-time monitoring and control of Critical Process Parameters (CPPs) and Critical Quality Attributes (CQAs) during manufacturing, essential for continuous processing [6]. |
| Scalable Lab System (SLS) Mill | A milling platform that allows for easy technology screening and direct scale-up from R&D to production by maintaining consistent particle size distribution (PSD) across scales [45]. |
Optimizing PMI and Overcoming Implementation Barriers
Identifying and Mitigating Major Cost Drivers (CAPEX/OPEX)
Frequently Asked Questions (FAQs)
1. What are the primary cost drivers (CAPEX vs. OPEX) for batch and continuous processes? The primary cost drivers differ significantly between the two modes. For batch processes, operational expenditures (OPEX) are often higher due to lower production rates, more frequent equipment cleaning and maintenance, higher labor costs, and increased energy consumption from frequent startups [4] [28]. For continuous processes, capital expenditure (CAPEX) is the major driver, requiring a significant initial investment in specialized, automated machinery and sophisticated control systems [4] [49] [22]. However, once established, continuous processing typically incurs lower OPEX through higher production rates, reduced labor, and lower unit costs [4] [28] [50].
2. From a PMI perspective, is a continuous process always more sustainable than a batch process? Not necessarily. Research indicates that the Process Mass Intensity (PMI) of a continuous manufacturing process for biologics can be comparable to that of batch processes [2]. While PMI is a useful benchmarking metric, it does not account for factors like energy consumption. A continuous process with a higher PMI might still be more environmentally sustainable overall if its productivity (in grams of drug substance per unit time) is multifold higher, leading to lower overall energy consumption per unit produced [2].
3. What are the common operational (OPEX) failures in continuous processing and how can they be mitigated? A common and costly failure in continuous operations is unplanned downtime due to equipment malfunction [4] [22]. Mitigation strategies include:
- Proactive Maintenance: Implementing advanced sensor technology and automated monitoring systems for real-time equipment health checks and predictive maintenance [28] [22].
- Skilled Labor: Investing in extensive training for technicians to operate and troubleshoot complex machinery, thereby reducing downtime [22]. Another challenge is the inflexibility to accommodate product variety, which can lead to costly changeovers [4] [49]. A thorough production volume analysis before implementation is key to ensuring continuous processing is the right fit [49].
4. How does production volume influence the decision between batch and continuous processing? Production volume is a critical factor. Batch processing is well-suited for small to medium volumes, niche markets, and products requiring customization [4] [49]. Continuous processing excels in large-scale production of standardized products, where its high throughput and operational efficiency lead to significantly lower costs per unit [4] [28] [49]. The table below summarizes key comparative data to guide this decision.
| Factor | Batch Process | Continuous Process |
|---|---|---|
| Typical Production Volume | Small to medium volumes [4] [28] | Large-scale, high-volume output [4] [28] |
| Relative CAPEX | Lower initial investment [49] [22] | Significantly higher initial investment [49] [22] |
| Relative OPEX | Higher unit costs (labor, maintenance, energy) [4] [28] | Lower unit costs at high volumes [4] [28] |
| Net Present Value (NPV) Attractiveness | Less attractive for new U.S. facilities compared to CM [50] | More attractive for new U.S. facilities for both brand and generic companies [50] |
| Process Mass Intensity (PMI) | Can be comparable to continuous processes for biologics [2] | Can be comparable to batch processes; overall sustainability requires broader metrics [2] |
5. What regulatory challenges are unique to continuous manufacturing in drug development? The regulatory landscape for continuous manufacturing is evolving. A key challenge is demonstrating consistent product quality through real-time monitoring and control throughout an uninterrupted production run [28]. Regulatory strategies must be adapted from traditional batch-based quality control, which relies on end-product testing [28]. Implementing Quality by Design (QbD) principles and advanced process analytical technology (PAT) is crucial for ensuring compliance and building regulatory confidence in the continuous process [51].
Troubleshooting Guides
Issue 1: High Operational Costs (OPEX) in Batch Processing
Symptoms:
- Rising costs per unit of production.
- Frequent equipment setup and cleaning leading to downtime.
- High energy bills from repeated process startups and shutdowns [4] [28].
Diagnosis and Resolution:
| Step | Action | Rationale & Reference |
|---|---|---|
| 1. Process Intensification | Explore technologies like flow chemistry or advanced bioreactors to create semi-continuous operations within a batch framework. | Increases productivity and reduces cycle time, directly improving material and energy efficiency [2]. |
| 2. PMI Analysis | Calculate the Process Mass Intensity for your batch process. | Identifies the largest contributors to material waste, allowing for targeted optimization of reagents and solvents [52]. |
| 3. Energy Audit | Conduct an audit of energy usage during different production stages. | Pinpoints inefficiencies related to heating, cooling, and agitation, enabling corrective measures [2] [4]. |
Issue 2: Managing High Capital Expenditure (CAPEX) in Continuous Processing
Symptoms:
- High upfront investment in automated equipment and control systems.
- Long payback periods on capital investment.
- Budget overruns during project implementation [49] [22].
Diagnosis and Resolution:
| Step | Action | Rationale & Reference |
|---|---|---|
| 1. Pilot Plant Testing | Conduct thorough testing at the pilot plant scale before full-scale investment. | Validates process feasibility and boundary conditions, de-risking the large capital investment [53]. |
| 2. Stochastic NPV Analysis | Perform a net present value (NPV) simulation that accounts for cost and revenue uncertainties. | Provides a robust economic analysis, demonstrating that for new U.S. facilities, CM is often more economically attractive than batch despite higher CAPEX [50]. |
| 3. Phased Implementation | Consider a semi-continuous process where only the most value-added steps are converted to continuous flow. | Reduces initial capital outlay while capturing some efficiency benefits, offering a compromise solution [28]. |
Issue 3: Quality Control Deviations in Continuous Operation
Symptoms:
- Inconsistent product quality over time.
- Difficulties in real-time monitoring and immediate correction of process deviations [4].
Diagnosis and Resolution:
| Step | Action | Rationale & Reference |
|---|---|---|
| 1. Implement PAT | Integrate Process Analytical Technology (PAT) tools such as inline sensors for real-time monitoring of critical quality attributes. | Enables immediate detection and correction of deviations, ensuring consistent quality and minimizing waste [28]. |
| 2. Automated Control Loops | Develop and validate automated feedback control systems to adjust process parameters in real-time. | Maintains the process within a predefined "state of control" without manual intervention, enhancing reliability [4] [22]. |
| 3. Enhanced Sampling | Establish rigorous sampling protocols at key points in the continuous process for offline analysis. | Provides supplementary data to validate real-time monitoring and ensures comprehensive quality oversight [28]. |
The Scientist's Toolkit: Key Research Reagent Solutions
The following table details essential materials and their functions relevant to experiments comparing batch and continuous processes.
| Reagent/Material | Function in Experimentation |
|---|---|
| Process Mass Intensity (PMI) Calculator | A tool to quantify the total mass of materials used per unit of product, enabling direct comparison of process efficiency and environmental impact [52]. |
| Inline Analytical Sensors (e.g., NIR, Raman) | Enable real-time monitoring of reaction conversion, product concentration, and critical quality attributes during a continuous process [28]. |
| Stable Isotope Tracers | Used to track material flow and residence time distribution in continuous systems, crucial for understanding and modeling process dynamics. |
| Customizable Pilot Plant System | A small-scale, flexible system that allows researchers to simulate and test both batch and continuous processes to gather comparative data on yield, productivity, and cost [53]. |
Experimental Workflow for Process Comparison
The diagram below outlines a high-level experimental workflow for conducting a comparative analysis of batch and continuous processes, incorporating key decision points.
Process Comparison Workflow
Strategies for Process Intensification to Reduce PMI
This technical support resource is framed within a broader research thesis comparing Process Mass Intensity (PMI) in batch versus continuous manufacturing processes. PMI, calculated as the total mass of inputs (e.g., solvents, water, reagents) per mass of product output, is a key metric for assessing process sustainability [54]. Process Intensification (PI) aims to make manufacturing processes significantly more efficient, compact, and sustainable, often through strategies like transitioning from batch to continuous processing and integrating unit operations [55]. This guide provides troubleshooting support for researchers and scientists implementing these strategies to reduce PMI in pharmaceutical development and manufacturing.
Core Concepts and Quantitative Framework
FAQ: What is PMI and why is it a crucial metric for Process Intensification?
A: Process Mass Intensity (PMI) is a sustainability metric that quantifies the total mass of materials used to produce a unit mass of a product, such as an Active Pharmaceutical Ingredient (API). It is calculated as [54]: PMI = Total Mass of Inputs (kg) / Mass of Product (kg) Inputs include solvents, water, reagents, and other raw materials. A lower PMI indicates a more efficient and less wasteful process. PI strategies target drastic reductions in PMI by redesigning processes to be more efficient, often by combining operations, improving heat and mass transfer, and moving to continuous manufacturing [55]. In the pharmaceutical industry, solvents and water are typically the largest contributors to PMI, making them primary targets for intensification efforts [54].
FAQ: How does continuous processing typically compare to batch processing in terms of PMI?
A: Research indicates that the PMI of continuous manufacturing processes can be comparable to that of batch processes for biologics like monoclonal antibodies [2]. However, PMI alone does not always capture the full sustainability picture. One study noted that a continuous process with a higher PMI could still be more environmentally sustainable than a batch process with a lower PMI if the continuous process has a much higher productivity (g of drug substance per unit time), leading to lower overall energy consumption per unit produced [2]. For small molecule APIs, continuous processing is often pursued specifically for its potential to lower PMI through higher product yields, more consistent quality, and easier scale-up [54].
Table: Key Quantitative Comparisons Between Batch and Continuous Processing
| Aspect | Batch Processing | Continuous Processing | Key PI Driver |
|---|---|---|---|
| Process Mass Intensity (PMI) | Can be high due to cleaning, purification, and lower yields [54] | Potential for significant reduction via streamlined, efficient operations [54] | Integration and efficiency |
| Capital Expenditure (Capex) | Traditional standard | Can be 20-76% lower (Oral Solid Dose case study) [56] | Modularity and compactness |
| Operational Savings | Traditional standard | Can be 9-40% lower (Oral Solid Dose case study) [56] | Reduced energy and material use |
| Energy Consumption | Can be significant, often from distillation/drying [54] | Potentially lower per unit of product, especially at high productivity [2] | Improved heat/mass transfer |
| Process Scalability | Scale-up can be complex and constrained by equipment [54] | Easier scale-out via numbering-up; constrained by chemistry/physics [54] | Modular and flexible design |
Technology Implementation and Troubleshooting
This section addresses common challenges and questions when implementing specific PI technologies to reduce PMI.
Technology Guide 1: Flow Chemistry and Microreactors
Q: We are experiencing challenges with fouling and clogging in our microreactors. What are the primary causes and solutions? A: Fouling and clogging are frequent challenges in continuous flow systems, often stemming from solid formation or precipitation.
- Potential Cause 1: Poor solubility of reactants or intermediates in the reaction stream.
- Troubleshooting: Increase solvent strength or use a co-solvent, ensuring compatibility with reactor materials. Consider increasing reactor temperature to enhance solubility, a feat more safely achieved in a flow system than batch [55].
- Potential Cause 2: Formation of particulates or gases as by-products.
- Troubleshooting: Implement in-line filters or ultrasound probes to disrupt particle aggregation. For gas-liquid reactions, use specific flow reactor designs that efficiently handle and separate multiphase streams.
- General Protocol: Before scaling, conduct a thorough solubility analysis of all reagents and potential intermediates under reaction conditions. Start with a conservative residence time and adjust.
Q: How do we control highly exothermic reactions in a flow reactor to improve safety and selectivity? A: The high surface-area-to-volume ratio of microreactors provides exceptional heat transfer capabilities, making them ideal for exothermic reactions.
- Experimental Protocol:
- Characterization: First, use reaction calorimetry in batch to quantify the heat release.
- Reactor Selection: Choose a microreactor (e.g., chip-based, tubular) with a high heat exchange coefficient.
- Temperature Control: Implement a control system where the reactor is embedded within a heat exchanger jacket. The high heat transfer allows for near-isothermal operation, suppressing hot spots and improving reaction selectivity [55] [54].
- Monitoring: Use in-line Process Analytical Technology (PAT) to monitor temperature and conversion in real-time.
Technology Guide 2: Integrated/Hybrid Unit Operations
Q: What are the key control challenges in reactive distillation columns, and how can they be managed? A: Reactive distillation integrates reaction and separation, intensifying the process but introducing complex, nonlinear dynamics that challenge traditional control systems [57].
- Challenge: Strong interaction between reaction and separation variables (e.g., temperature, composition, flow rates) can lead to complex dynamic behavior that is difficult for simple PID controllers to manage.
- Solution: Implement Advanced Process Control (APC) strategies.
- Model Predictive Control (MPC): This is a preferred advanced strategy. MPC uses a dynamic model of the column to predict future behavior and optimize multiple control variables simultaneously while respecting process constraints. It is particularly effective for handling the multivariable interactions in intensified systems like reactive distillation [57].
- Protocol for Implementation:
- Develop a Model: Create a first-principles or data-driven dynamic model of the reactive distillation process.
- Design Controller: Configure the MPC to control key parameters like product purity and conversion rate by manipulating inputs like reflux ratio and reboiler duty.
- Validate and Commission: Test the control strategy offline via simulation (e.g., using a digital twin) before deploying it on the physical system [57].
The following diagram illustrates a recommended control and optimization workflow for an intensified process unit like reactive distillation.
Diagram: Control Strategy Workflow for Intensified Units. This outlines the development and deployment of advanced control for complex PI systems like reactive distillation.
Advanced Strategies and Sustainability Integration
FAQ: Beyond new hardware, what synthetic chemistry strategies can reduce PMI?
A: Several chemistry-led strategies can dramatically reduce material usage and waste.
- Catalysis: Employing catalytic processes (chemical, enzymatic) reduces the stoichiometric quantities of reagents needed, minimizing waste [54]. Biocatalysts are especially attractive due to their high selectivity, often eliminating the need for protection/deprotection steps and their associated solvents and reagents [54].
- One-Pot and Multicomponent Reactions (MCRs): Conducting multiple synthetic steps in a single reactor without intermediate isolation minimizes purification challenges and solvent use, leading to high atom economy and lower PMI [54].
- Alternative Energy Inputs: Using microwave irradiation, ultrasound, or photochemistry can activate reactions more selectively and efficiently, potentially speeding up reactions and reducing solvent use [55] [54]. Photochemistry, especially when coupled with flow reactors, allows for precise light delivery and control over reactive intermediates.
Scientist's Toolkit: Key Research Reagent Solutions for PI
Table: Essential materials and technologies for developing intensified processes with lower PMI.
| Tool/Reagent | Function in Process Intensification | Key Consideration for PMI Reduction |
|---|---|---|
| Heterogeneous Catalysts | Provides active sites for reaction; easily separated and reused. | Reduces need for stoichiometric reagents, lowering mass input and waste [54]. |
| Immobilized Enzymes | Biocatalysts offering high selectivity under mild conditions. | Avoids protection groups, reduces steps, and enables greener solvents, cutting PMI [54]. |
| Supported Reagents | Reagents immobilized on a solid support. | Simplifies work-up and purification, reducing solvent use in separation steps [54]. |
| Process Analytical Technology (PAT) | Sensors for real-time monitoring of process parameters (e.g., concentration, pH). | Enables real-time adjustments to maintain optimal yield and minimize off-spec material [56]. |
| Digital Twin Software | Virtual replica of the physical process for simulation and optimization. | Allows for virtual testing of PI strategies to predict their impact on PMI before costly experiments [57] [56]. |
FAQs
1. How do equipment design requirements differ fundamentally between batch and continuous processes for PMI comparison studies?
The core difference lies in integration and operation mode. Batch processing uses discrete, standalone unit operations with defined start and end points, requiring equipment for handling, storing, and monitoring intermediate products between each phase [6]. Conversely, continuous manufacturing involves a single, fully integrated production line where material flow is uninterrupted [6] [27]. Equipment for continuous processes is designed to operate 24/7, is often smaller, and is located within a single facility, eliminating the need for intermediate storage and transport equipment [27] [11].
2. What specific material properties pose the greatest challenge when designing equipment for continuous manufacturing?
The flow properties of powders and the behavior of solids in flow reactors are significant challenges. For powder blending in continuous oral solid dosage (OSD) production, material properties like powder flow profiles have a minimal impact on blend uniformity, which is an advantage over batch processes [6]. However, in flow reactors for active pharmaceutical ingredient (API) synthesis, solid materials can cause blockages in reactor pipes. Furthermore, stainless steel equipment may be unsuitable for corrosive acidic products, necessitating costly machinery upgrades [11].
3. In continuous processing, how is quality control for material properties achieved differently than in batch?
Quality control in batch manufacturing typically relies on end-point testing, where intermediate products are collected and evaluated before proceeding to the next phase [6] [58]. In continuous manufacturing, a robust control strategy using Process Analytical Technology (PAT) is essential for real-time monitoring and control [6] [27] [58]. PAT tools and sensors monitor Critical Process Parameters (CPPs) and Critical Quality Attributes (CQAs) throughout the uninterrupted process, enabling immediate corrections and ensuring material properties remain within specifications [27] [58].
4. What are the common points of failure in integrated continuous equipment, and how can they be mitigated?
A common challenge is the failure or required adaptation of a single unit operation, which can disrupt the entire integrated line [58]. A proposed mitigation strategy is a modular two-layer control system. Instead of one complex controller for the entire line, an upper layer manages overall system targets, and a separate unit operation control layer determines manipulated variables for each step. This structure allows the system to maintain production even if one unit's controller fails or needs adaptation [58]. Additionally, equipment overheating or material accumulation during longer runs are potential failures that require design consideration [6].
Troubleshooting Guides
Problem 1: Powder Feed Blockages in Continuous Oral Solid Dosage Lines
| Step | Action | Technical Rationale |
|---|---|---|
| 1. Immediate Response | Pause the feeder and safely isolate the material hopper. | Prevents further pressure buildup and potential damage to the feeding mechanism. |
| 2. Diagnosis | Inspect the feed hopper and screw auger for bridging or cohesive powder. | Identifies the root cause, such as poor flowability due to static charge or moisture. |
| 3. Resolution | Carefully clear the blockage. Implement powder preconditioning (e.g., milling, dehumidification). | Restores flow. Addresses the material property issue to prevent recurrence. |
| 4. Verification | Re-calibrate the feeder and conduct a short run with PAT monitoring of blend uniformity. | Ensures the system is operational and that the corrective action has not adversely affected product quality. |
Problem 2: Inconsistent Product Quality at Bioreactor Outlet in Continuous Biologics Processes
| Step | Action | Technical Rationale |
|---|---|---|
| 1. Monitoring Check | Verify the calibration and function of all PAT probes (e.g., for pH, dissolved oxygen, metabolite concentration). | Ensures that the data used for process control is accurate and reliable. |
| 2. Process Parameter Audit | Review historical data for CPPs like residence time, temperature, and feed composition for deviations. | Identifies process drifts that may be causing the variation in a Critical Quality Attribute (CQA). |
| 3. Control Strategy | Implement or adjust a feedback control loop to manipulate a key CPP (e.g., nutrient feed rate) based on the real-time CQA measurement. | Actively corrects the process to bring the product quality back to its target value. |
| 4. Sampling & Analysis | Take a small sample for off-line analysis to cross-verify the PAT data and cell viability. | Provides a definitive quality measurement and helps diagnose potential PAT sensor drift. |
Experimental Protocols
Protocol 1: Comparative PMI Assessment for a Model API
1. Objective: To quantitatively compare the Process Mass Intensity (PMI) of batch versus continuous manufacturing processes for a specific model API, such as (2-phenylcyclopropyl)methanol synthesized via a Simmons-Smith reaction [58].
2. Materials:
- Raw materials for API synthesis (e.g., cinnamyl alcohol, reagents for Simmons-Smith reaction)
- Solvents for purification (for extraction and washing)
- Batch manufacturing equipment (reactor, filter dryer, purification columns)
- Integrated continuous manufacturing line (continuous flow reactor, solid-liquid separation unit, liquid-liquid extraction unit) [58]
- Analytical instruments (HPLC, balances)
3. Methodology:
- Batch Process: Execute the synthesis and purification in discrete, sequential steps. Record the mass of all input materials (raw materials, solvents, etc.) consumed for a single batch. Measure the mass of the final Drug Substance (DS) produced [2].
- Continuous Process: Run the integrated continuous line at a steady state for a defined period. Continuously record the mass flow rates of all input streams. Collect and measure the total mass of DS produced over that period [58].
- Calculation: For both processes, calculate the PMI using the formula: PMI = Total Mass of Input Materials (kg) / Mass of Drug Substance (kg) [2] [58].
Protocol 2: Real-Time Quality Control Using PAT in Continuous Blending
1. Objective: To demonstrate real-time monitoring and control of blend homogeneity in a continuous direct compression line for pharmaceutical tablets.
2. Materials:
- Continuous powder blender (e.g., gravimetric feeder with integrated mixer)
- API and excipients
- PAT tool (e.g., NIR spectrometer) installed at the blender outlet
- Data acquisition and control system
3. Methodology:
- Setup: Calibrate the NIR spectrometer to correlate spectral data with API concentration.
- Operation: Start the continuous blender, feeding API and excipients at predetermined rates.
- Monitoring & Control: The NIR sensor continuously measures the API concentration in the flowing powder blend. This real-time data is fed into a process control system. If the measured concentration deviates from the set point, the control system automatically adjusts the feeder rates of the API or excipients to correct the blend uniformity without stopping the process [27] [58].
- Verification: Periodically collect small samples for off-line analysis to validate the accuracy of the PAT data.
Quantitative Data Comparison
Table 1: Operational and Environmental Comparison of Batch vs. Continuous Processes
| Parameter | Batch Process | Continuous Process | Source |
|---|---|---|---|
| Production Time | Weeks to months | Can be reduced to a single day | [11] |
| Process Mass Intensity (PMI) for mAbs | Comparable to continuous | Comparable to batch | [2] |
| Relative Energy Consumption | Higher per unit DS (in specific cases) | Can be lower per unit DS due to higher productivity | [2] |
| Scale-up Method | Sequential batch runs | Increased run time or equipment size adjustment | [6] [27] |
| Facility Footprint | Larger, often multiple facilities | Smaller, single facility | [27] [11] |
Table 2: Quality Control and Operational Efficiency Metrics
| Parameter | Batch Process | Continuous Process | Source |
|---|---|---|---|
| Quality Control Approach | Off-line, end-point testing | Real-time monitoring (PAT) | [6] [58] |
| Impact of Quality Failure | Rejection of entire batch | Rejection of a limited product quantity | [6] |
| Manufacturing Cost Reduction | Baseline | Variable costs reduced by 40-50% | [11] |
| Equipment Flexibility | High, reconfigurable for different products | Low, specialized for a specific product | [28] [4] |
Research Reagent Solutions
Table 3: Essential Materials for Batch vs. Continuous PMI Experiments
| Item | Function | Application Note |
|---|---|---|
| Model API Substrate (e.g., Cinnamyl Alcohol) | The starting material for API synthesis; its properties affect reaction kinetics and purification. | Used in the Simmons-Smith reaction for a model API to ensure a direct comparison between processes [58]. |
| Process Analytical Technology (PAT) Probes | Sensors for real-time monitoring of Critical Process Parameters (CPPs) and Critical Quality Attributes (CQAs). | Essential for quality control in continuous processing; e.g., NIR for blend uniformity, pH and DO probes in bioreactors [27] [58]. |
| Certified Reference Materials (CRMs) | Calibrate analytical instruments (XRF, OES) for accurate Positive Material Identification of alloy components. | Ensures the accuracy and reliability of material verification for process equipment, crucial for safety and compliance [59]. |
| High-Performance Alloys | Materials for constructing reactors and pipes that resist corrosion and handle high-pressure flow chemistry. | Critical for continuous equipment design, especially when processing corrosive acidic products or APIs [11]. |
Process Workflow and Control Diagrams
Navigating Organizational and Regulatory Barriers to Adoption
The transition from traditional batch manufacturing to continuous manufacturing (CM) represents a significant paradigm shift in the pharmaceutical industry. While research consistently shows that Continuous Manufacturing offers substantial benefits, including a 70-90% reduction in manufacturing time and a 25-50% decrease in energy and water consumption, its widespread adoption faces significant organizational and regulatory hurdles [32]. This technical support center is designed within the context of a broader thesis comparing the Process Mass Intensity (PMI) of batch versus continuous processes. It aims to provide researchers, scientists, and drug development professionals with practical troubleshooting guides and FAQs to navigate the complex implementation landscape, leveraging the most current data and regulatory insights.
Key Quantitative Comparison: Batch vs. Continuous Manufacturing
The following table summarizes core quantitative benefits of Continuous Manufacturing over batch processes, which serve as a powerful driver for adoption despite existing barriers [32].
| Performance Metric | Improvement in Continuous vs. Batch Manufacturing |
|---|---|
| Manufacturing Time | Reduced by 70-90% [32] |
| Production Efficiency | Improved by up to 90% [32] |
| Product Quality Standard Attainment | Over 95% of products meet quality standards [32] |
| Dose Uniformity | Improved by 40% [32] |
| Equipment/Plant Footprint | Requires 30-50% less space [32] |
| Energy and Water Consumption | Reduced by 25-50% [32] |
Troubleshooting Common Adoption Barriers
Regulatory and Compliance Hurdles
Q1: What are the most common regulatory concerns when submitting a Continuous Manufacturing process for approval?
Regulatory uncertainties often stem from the integration of novel technologies and differing system dynamics compared to batch processing [32] [27]. Key concerns for regulators include:
- Material Traceability: Ensuring the ability to track material through a continuous, integrated system [32].
- Process Control and Monitoring: Defining and validating Real-Time Release Testing (RTRT) and the use of Process Analytical Technology (PAT) for continuous quality assurance [27].
- Batch Definition: Defining what constitutes a "batch" in a continuous process for the purpose of quality control and recall [32].
- Handling of Process Upsets: Demonstrating robust control strategies for detecting and diverting non-conforming material in real-time [32].
Q2: What strategic approach can mitigate regulatory risks during development?
Adopt a holistic Quality by Design (QbD) framework coupled with a robust Process Analytical Technology (PAT) strategy [27]. This involves:
- Proactively identifying Critical Material Attributes (CMAs), Critical Process Parameters (CPPs), and Critical Quality Attributes (CQAs) during development.
- Implementing advanced process controls that use PAT data to automatically adjust CPPs to maintain CQAs within the desired state.
- Developing a comprehensive control strategy that includes a definition for the batch based on Residence Time Distribution (RTD) models and clear protocols for startup, steady-state operation, and shutdown [27].
Organizational and Cultural Challenges
Q3: Our organization is resistant to moving away from proven batch systems. How can we build internal support?
Organizational resistance is a significant barrier, often described as a "glass ceiling" that prevents project management and innovative processes from gaining strategic traction [60]. To overcome this:
- Demonstrate Tangible Value: Move beyond theoretical benefits and present pilot-scale data. Highlight the comparable Process Mass Intensity (PMI) and the potential for lower overall energy consumption per unit of drug substance produced, which speaks to both cost and sustainability goals [26].
- Secure Early Management Buy-in: The active involvement of top management in remediation and change is critical. Leaders must establish a strong governance framework that prioritates compliance and strategic alignment, and allocate sufficient resources for training and infrastructure [61].
- Reframe the Narrative: Position CM not as a mere technical change but as a strategic capability that enhances agility, flexibility, and response capacity during public health emergencies and supply chain disruptions [32] [27].
Q4: What are the typical internal structural barriers to adopting CM?
Based on project management glass ceiling research, barriers within the direct control of business include [60]:
- Pipeline Barriers: A lack of career development and recruitment policies that create opportunities for CM experts to advance.
- Organizational Structure: Structures that prevent direct reporting and communication between technical teams and senior strategic management.
- Corporate Climate: A culture that isolates technical teams from management roles and views CM as a tactical, rather than strategic, function.
Technical and Operational Obstacles
Q5: In a direct compression study, how did the variability of batch and continuous processes compare?
A detailed 2023 study comparing batch and continuous direct compression using similar tablet press setups found key differentiators [62]:
- The continuous process demonstrated significantly lower variability in tablet mass and tensile strength due to more consistent flow dynamics throughout the run.
- The batch process showed less consistent flow, leading to higher variability within the tablet press feed frame.
- However, the batch process exhibited a more consistent API concentration variability, attributed to its better-controlled (offline) blending procedure prior to tableting.
Q6: What is a critical material science consideration when designing a formulation for CM?
The flow dynamics in the operating system are a crucial differentiator. Material properties related to flow, compressibility, and permeability play a more significant role in continuous processes than in batch [62]. Therefore, excipient selection is paramount, and a thorough raw material characterization is a non-negotiable part of the experimental protocol.
Experimental Protocols and Methodologies
Protocol: Comparative Evaluation of Direct Compression Processes
This protocol is adapted from a study that directly compared batch and continuous direct compression using similar equipment, allowing for an accurate assessment of processability and final tablet quality [62].
1. Objective: To compare the overall material processability and final tablet quality of formulations processed via batch and continuous direct compression, and to correlate material properties and process parameters with tablet properties.
2. Materials:
- API: Paracetamol Powder (or model API with intermediate flow and cohesion properties).
- Fillers: A variety of lactose-based fillers (e.g., Spray-dried, Anhydrous, Granulated) and Microcrystalline Cellulose.
- Excipients: Superdisintegrant (e.g., Sodium Starch Glycolate), Lubricant (e.g., Magnesium Stearate).
3. Equipment:
- Continuous Line: Integrated continuous direct compression (CDC) line comprising feeders, a continuous blender, and a rotary tablet press.
- Batch Line: A stand-alone rotary tablet press (configured similarly to the continuous line press) fed by a hopper with a rotating valve to simulate batch feeding.
4. Method Formulation:
- Prepare two primary formulation types:
- Low-Dosed: 1% w/w API.
- High-Dosed: 40% w/w API.
- Keep disintegrant (4% w/w) and lubricant (1% w/w) concentrations constant.
- Process each formulation in both the batch and continuous systems.
5. Data Collection and Analysis:
- Processability Metrics: Monitor feeder mass flow consistency and tablet press feed frame stability.
- Tablet Quality Metrics: Measure tablet weight uniformity, tensile strength, and API content uniformity at regular intervals throughout the run.
- Multivariate Analysis: Use Partial Least Squares (PLS) regression to link raw material properties and process parameters to the final tablet quality responses for both processes.
Workflow Diagram: Batch vs. Continuous Manufacturing
The diagram below illustrates the fundamental operational differences between batch and continuous direct compression processes, highlighting the streamlined nature of CM.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key materials used in comparative studies of direct compression, along with their critical function in the process [62].
| Material Name | Function / Critical Role | Key Characteristics for CM |
|---|---|---|
| Spray-Dried Lactose (e.g., SuperTab 11SD) | Filler/Diluent | Good flowability and compressibility; crucial for consistent feeding and blending. |
| Anhydrous Lactose (e.g., SuperTab 22AN) | Filler/Diluent | Different compaction behavior compared to other lactoses; impacts tablet tensile strength. |
| Microcrystalline Cellulose (e.g., Pharmacel 102) | Filler/Binder | Excellent compressibility; often used in blends to improve the compactability of other fillers. |
| Silicified MCC (e.g., Pharmacel sMCC90) | Filler/Binder | Enhanced flow and compaction properties compared to standard MCC. |
| Sodium Starch Glycolate (e.g., Primojel) | Superdisintegrant | Ensures tablet disintegration; concentration must be optimized for formulation. |
| Magnesium Stearate | Lubricant | Reduces friction during compression; over-lubrication can negatively impact tablet strength. |
| Paracetamol Powder | Model API | Used in studies for its intermediate flow and cohesion properties, making it a good challenge. |
Leveraging Pharma 4.0 and Real-Time Monitoring for Efficiency
Technical Support Center
Troubleshooting Common Implementation Issues
Issue 1: Poor Data Integration Between Legacy and New Systems
- Problem: New monitoring sensors are installed, but data is not flowing correctly to the central platform, creating information silos.
- Solution: Implement a middleware or industrial data hub that supports standard communication protocols (e.g., OPC-UA). Start with a pilot project on a single process unit to map all data points and ensure integrity before full-scale rollout [63].
Issue 2: Real-Time Monitoring Alarms are Overwhelming Operators
- Problem: The system generates excessive false alarms or minor alerts, leading to alarm fatigue and missed critical events.
- Solution: Reclassify alarms based on risk assessment. Implement alarm filtering and grouping logic. Use a phased approach: start with critical process parameters (CPPs) only, then gradually incorporate other data points as operators adapt [64] [65].
Issue 3: PAT Tools Provide Inconsistent Readings in Continuous Processes
- Problem: Spectroscopic tools (e.g., NIR, Raman) used for real-time quality attribute measurement show drift or noise during long production runs.
- Solution: Establish a robust calibration and validation schedule. For continuous processes, implement automated recalibration routines during scheduled maintenance windows. Use redundant sensors for critical measurements to cross-verify data [64] [6].
Frequently Asked Questions (FAQs)
Q1: What is the most significant operational difference when implementing real-time monitoring in batch versus continuous manufacturing? The key difference lies in quality control timing and response. In batch processes, monitoring and corrective actions occur at the end of each discrete step. In continuous processes, monitoring and adjustments must happen in real-time throughout the uninterrupted production flow, as any discrepancy can affect the entire production run [6] [28].
Q2: How does the role of the operator change with Pharma 4.0 implementation? The role evolves from manual execution and documentation to system supervision and data-driven decision-making. Pharma 4.0 empowers operators by automating high-risk and repetitive tasks, providing real-time insights for intervention, and reducing human error. The focus shifts to exception handling and process optimization [66].
Q3: What are the first steps in retrofitting a legacy batch line for Pharma 4.0 capabilities? Begin with a comprehensive assessment of current infrastructure and data maturity. Prioritize investments in non-invasive, rapid PAT tools (like Raman spectroscopy) and IoT-enabled sensors for critical process parameters. Focus initial integration on a modular, scalable platform that can grow with your digital transformation roadmap [64] [63].
Quantitative Comparison: Batch vs. Continuous Manufacturing
Table 1: Performance and Operational Comparison
| Aspect | Batch Manufacturing | Continuous Manufacturing |
|---|---|---|
| Production Rate & Volume | Slower, limited by batch size and handling times [28] | Higher speed and volume through 24/7 operation [28] [67] |
| Operational Flexibility | High; easy to modify recipes and equipment between runs [28] [67] | Low; specialized equipment designed for a specific product [28] |
| Quality Control Approach | Testing at the end of each batch step [28] [67] | Real-time monitoring with PAT and sensors [6] [28] |
| Primary Cost Driver | Higher unit costs from lower rates and more frequent cleaning [28] | Lower unit costs from higher efficiency and scale, but requires significant initial investment [6] [28] |
| Typical Maintenance | Periodic, between production runs [28] | Predictive, based on real-time equipment monitoring to prevent downtime [67] |
Table 2: Impact of Pharma 4.0 and Real-Time Monitoring
| Metric | Batch Manufacturing | Continuous Manufacturing |
|---|---|---|
| Quality Control Time | Reduced through automated data collection [64] | Reduced by 50-70% [6] |
| Process Validation | Based on completed batch records [63] | Shifts to continuous process verification [63] |
| Response to Deviation | Corrective actions between batches [28] | Immediate adjustments during production [64] [6] |
| Risk from Non-Conformity | Rejection of an entire batch [6] | Rejection of a limited product quantity [6] |
Experimental Protocol: Implementing a Real-Time Monitoring Workflow
This protocol outlines the methodology for integrating real-time monitoring to track a Critical Process Parameter (CPP) and its effect on a Critical Quality Attribute (CQA).
1. Definition of Critical Elements
- Identify CQAs: Define the specific product quality characteristics (e.g., concentration, particle size, purity) that must be controlled within predefined limits [64].
- Identify CPPs: Determine the process parameters (e.g., temperature, pressure, flow rate) that directly influence the identified CQAs [64].
2. Sensor and PAT Tool Selection & Calibration
- Selection Criteria: Choose non-destructive, non-invasive sensors (e.g., NIR, Raman spectroscopy) that provide rapid, actionable data suitable for the production environment [64].
- Calibration: Develop a calibration model by correlating sensor output with traditional lab-based measurements of the CQA across a range of expected operating conditions [64].
3. System Integration and Data Architecture
- Connectivity: Integrate sensors into the network using IoT protocols for seamless data flow [66] [65].
- Data Platform: Feed data into a central platform (e.g., ValGenesis Process Insight) for centralized, real-time viewing of CPPs and CQAs. Automate data collection and report generation [64].
- Control Logic: Establish automated feedback loops or alerts that trigger when CPPs deviate from their set ranges, enabling immediate corrective actions [64].
4. Workflow Execution and Continuous Verification
- Execution: Run the process, allowing the system to continuously monitor and control the CPP.
- Verification: Periodically collect manual samples to verify the accuracy of the real-time PAT data, ensuring the model's reliability throughout the production run [64] [63].
The diagram below illustrates this integrated workflow.
Real-Time Monitoring Control Workflow
The Researcher's Toolkit: Essential Technologies for Pharma 4.0
Table 3: Key Research Reagent Solutions & Technologies
| Tool/Technology | Primary Function | Relevance to Batch/Continuous Research |
|---|---|---|
| PAT Tools (NIR, Raman) | Non-invasive, rapid analysis of CQAs during processing [64]. | Critical for continuous process control; enhances batch understanding [64] [6]. |
| IoT-Enabled Sensors | Collect real-time data on CPPs (T, pH, pressure) [66] [65]. | Foundational for data-driven research in both batch and continuous modes [66]. |
| Cloud-Based Data Platforms | Centralized repository for real-time and historical process data [64] [65]. | Enables advanced analytics and comparison of batch vs. continuous datasets [68]. |
| Electronic Quality Management System (eQMS) | Digital management of quality events, deviations, and documentation [63]. | Supports the rigorous data integrity and compliance needs for process comparison studies [63]. |
| Digital Twin | A virtual replica of the manufacturing process for simulation [64]. | Allows for low-risk optimization and scenario testing before physical experiments [64]. |
A Direct Comparison: Validating PMI and Economic Performance
Quantitative Comparison: Batch vs. Continuous Processes
The following tables summarize the key quantitative and qualitative differences between batch and continuous manufacturing processes to aid in research and process selection.
Table 1: Production Characteristics and Economic Factors
| Factor | Batch Process | Continuous Process |
|---|---|---|
| Production Volume | Small to medium volumes; suitable for ~74% of chemicals produced at <1,000 tons/year [44] | Large-scale, high-tonnage output; e.g., 157M tons/year of polyethylene [44] |
| Production Rate | Slower due to start/stop cycles; limited by batch capacity [28] | Higher, constant production rate; 24/7 operation minimizes bottlenecks [28] [67] |
| Unit Cost | Higher unit costs [28] | Lower unit costs due to higher output and efficiency [28] |
| Initial Investment | Lower initial setup cost [4] | Significant initial investment for specialized equipment [4] [69] |
| Operational Cost | Higher from frequent setup, cleaning, and energy for start/stops [28] [4] | Lower cleaning/maintenance costs per unit; better energy efficiency at steady-state [28] [44] |
| Economic Breakeven | Economically viable at lower volumes and for niche markets [4] | Requires high-volume production; suitable for plants running at >80% capacity [44] |
Table 2: Process Control, Quality, and Flexibility
| Factor | Batch Process | Continuous Process |
|---|---|---|
| Process Flexibility | High; equipment can be reconfigured for different products [28] [67] | Low; equipment specialized for a single product line [28] [4] |
| Customization | Ideal for products requiring customization or diverse portfolios [4] [69] | Ideal for standardized, single products with stable demand [28] [4] |
| Quality Control (QC) | QC at the end of each batch or processing step [28] [67] | Real-time, in-line monitoring using Process Analytical Technology (PAT) [69] |
| Product Consistency | Potential for batch-to-batch variation [69] | High consistency due to steady-state homogeneous conditions [28] [44] |
| Primary Industry Use | Dominant in pharmaceuticals, specialty/fine chemicals (85%), nanomaterials, food (e.g., baked goods) [28] [44] [69] | Dominant in commodity chemicals (e.g., ammonia, PVC), oil refining, metal fabrication [28] [44] |
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: Why does the batch process still dominate pharmaceuticals and nanomaterials, despite the known efficiency of continuous processing?
The reliance on batch processing is not due to outdated practices but is driven by practical, scientific, and economic factors [44]. Key reasons include:
- Low Production Volumes: An estimated 74% of chemical compounds (including many pharmaceuticals and nanomaterials) are produced in volumes of less than 1,000 metric tons per year, making continuous processing less economically viable [44].
- Precision and Control: Nanomaterials and pharmaceuticals require absolute precision during nucleation and growth phases. Batch processing provides a controlled environment where parameters like temperature and pH can be meticulously defined for each stage [44].
- Flexibility for R&D: Batch processes are inherently better suited for producing a diverse range of compounds and allow for easier scale-up or scale-out in response to uncertain market demand [44].
Q2: What are the primary cost-related trade-offs when choosing between batch and continuous methods?
The trade-offs are primarily between initial capital expenditure and long-term operational costs.
- Batch Processing has a lower initial investment but results in higher per-unit costs due to lower production rates, more frequent equipment cleaning, and energy consumption from repeated start-up cycles [28] [4].
- Continuous Processing requires a high initial capital outlay for specialized equipment and automation. However, once operational, it benefits from lower per-unit costs, reduced labor, and better energy efficiency during continuous steady-state operation [28] [44].
Q3: How does quality control differ fundamentally between the two processes?
The timing and methodology of quality control are fundamentally different.
- In Batch processing, quality is typically assessed at the end of a production run or after discrete steps. This allows for adjustments before the next batch but risks the loss of an entire batch if a fault is detected late [28] [69].
- In Continuous processing, quality control is integrated into the production line. Using Process Analytical Technology (PAT), critical quality attributes are monitored in real-time, allowing for immediate adjustments and ensuring consistent product quality throughout the production run [28] [69].
Troubleshooting Common Experimental and Process Issues
Issue 1: Inconsistent Results Between Batches in Batch Processing
- Potential Cause: Minor, unaccounted-for variations in processing times, temperature ramp rates, or raw material handling between batches [69].
- Solution:
- Standardize Protocols: Implement and strictly adhere to detailed Standard Operating Procedures (SOPs) for every step.
- Enhanced In-process Monitoring: Introduce more frequent and precise monitoring of critical parameters (e.g., temperature, pH, pressure) during, not just after, the batch process.
- Raw Material Analysis: Increase the scrutiny of raw material quality and consistency from different suppliers or lots.
Issue 2: Challenges in Achieving and Maintaining Steady-State in Continuous Processes
- Potential Cause: Fluctuations in feed stock composition, flow rates, or equipment performance (e.g., filter fouling, pump drift) preventing the system from reaching a stable equilibrium.
- Solution:
- Pre-conditioning: Ensure all feed stocks and reagents are pre-conditioned (e.g., temperature, concentration) before introduction to the system.
- Automated Control Loops: Implement robust, automated feedback control systems that can make fine adjustments to process parameters in response to sensor data from PAT tools.
- Preventive Maintenance: Establish a rigorous preventive maintenance schedule for all system components, especially pumps and sensors, to prevent performance decay [28].
Issue 3: System-Wide Failure in an Integrated Continuous Process
- Potential Cause: A failure in one unit operation (e.g., a clogged line in a purification step) can halt the entire integrated production train.
- Solution:
- Design for Redundancy: Where possible, incorporate parallel or backup units for critical steps that are prone to failure.
- Surge Capacity/Buffers: Introduce small, controlled surge tanks or buffers between major unit operations. This can decouple the steps, allowing one section to be temporarily stopped for maintenance without shutting down the entire line.
- Advanced Process Control (APC): Utilize APC systems that can predict potential failures based on trend analysis and initiate controlled shutdowns or process adjustments.
Experimental Protocols for Process Comparison
Protocol 1: Bench-Scale Batch Reaction Experiment
1.0 Objective: To execute a standard chemical synthesis (e.g., a simple esterification or hydrolysis) using a bench-scale batch reactor and characterize the output.
2.0 Materials and Equipment:
- Jacketed glass batch reactor (250 mL - 1 L)
- Overhead stirrer with torque control
- Heating/cooling circulator
- Temperature probe
- pH probe
- Sampling port
- Reagents as required by the specific reaction (e.g., substrates, catalysts, solvents)
3.0 Methodology:
- Charge: Add all initial reagents to the reactor according to the defined recipe.
- Initiate: Start stirring and begin the temperature ramp to the target reaction temperature. Record this as time zero (t=0).
- Monitor and Sample: At pre-defined time intervals (e.g., every 15 minutes), extract small samples via the sampling port.
- Quench and Analyze: Immediately quench each sample and analyze it using an appropriate method (e.g., HPLC, GC) to determine conversion and yield.
- Terminate: Once the target conversion is reached or the reaction is complete, actively cool the reactor to stop the reaction.
- Work-up: Transfer the entire batch for downstream work-up and purification as a single unit.
4.0 Data Analysis:
- Plot conversion and yield versus time.
- Calculate key performance indicators (KPIs) such as total cycle time, maximum yield, and volumetric productivity.
Protocol 2: Mini-Plant Continuous Flow Reaction Experiment
1.0 Objective: To execute the same chemical synthesis as in Protocol 1 using a continuous flow reactor and characterize the output at steady-state.
2.0 Materials and Equipment:
- Tubular continuous flow reactor (e.g., coiled tubing in a heated bath) or a packed-bed microreactor
- Two or more precision syringe or HPLC pumps for reagent feeds
- Heated chamber or oil bath for temperature control
- In-line pressure sensor and regulator
- Back-pressure regulator (BPR)
- In-line analytical probe (e.g., FTIR, UV) or automated fraction collector for off-line analysis
3.0 Methodology:
- System Priming: Prime the entire flow path with solvent and ensure the system is at the target temperature and pressure.
- Initiate Flow: Start the pumps simultaneously at the calculated flow rates to achieve the desired residence time.
- Steady-State Attainment: Allow the system to run, monitoring the output. Steady-state is achieved when key output parameters (e.g., conversion, pH) remain constant over time (typically 3-5 residence times).
- Steady-State Sampling: Once at steady-state, collect product output for a defined period for analysis.
- Parameter Variation: Systematically vary one parameter (e.g., residence time, temperature) while holding others constant, and repeat steps 3 and 4 to map the process window.
- Shutdown: Flush the system with a clean solvent.
4.0 Data Analysis:
- Record the steady-state conversion and yield for each set of parameters.
- Calculate KPIs like space-time yield and compare them to the batch KPIs from Protocol 1.
Process Workflow Visualization
Batch vs Continuous Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials and Equipment for Process Development
| Item | Function in Research |
|---|---|
| Single-Use Bioreactor/Reactor Assembly | Disposable bag-based systems for cell culture or chemical reaction; eliminates cleaning validation and cross-contamination between batches, ideal for flexible, multi-product facilities [69]. |
| Process Analytical Technology (PAT) | A system for real-time monitoring of Critical Quality Attributes (CQAs); includes in-line sensors (pH, concentration) and instruments (FTIR, UV) essential for controlling continuous processes [69]. |
| Precision Pumps (Quaternary Diaphragm, Syringe) | Provide consistent, pulseless flow of reagents in continuous processes; critical for maintaining accurate residence times and steady-state conditions [69]. |
| Single-Use Assemblies & Connectors | Pre-sterilized, integrated fluid pathways for transferring media, buffers, and product between unit operations; enable aseptic processing and system flexibility [69]. |
| Back-Pressure Regulator (BPR) | A crucial device in continuous flow systems that maintains a constant, elevated pressure within the reactor, preventing the formation of gas bubbles and ensuring consistent fluid properties and reaction rates. |
Analysis of Production Volume Impact on Cost and PMI
This guide provides technical support for researchers and scientists investigating the impact of production volume on cost and Process Mass Intensity (PMI) within pharmaceutical manufacturing. The core of this research often involves a direct comparison between traditional batch and modern continuous processing methods.
Batch Process Manufacturing is defined by producing discrete, finite quantities of product. Each batch must complete a full production step before the entire batch can move to the subsequent step [67] [29]. Continuous Process Manufacturing involves an uninterrupted flow of materials, where raw materials are continuously fed into the system and finished products emerge from the other end [67] [28]. Process Mass Intensity (PMI) is a key metric for assessing the sustainability of a process, calculated as the total mass of materials used to produce a unit mass of the active pharmaceutical ingredient (API) or drug substance [2]. Cost of Goods Sold (COGS) Variance analysis is crucial for understanding financial performance, breaking down cost differences into volume, mix, and rate components [70].
Troubleshooting Guides
High Process Mass Intensity (PMI)
Problem: The calculated PMI for a new process is unacceptably high, indicating poor material efficiency.
Solution: Investigate and optimize process parameters and technology.
- Check Underlying Assumptions: Do not rely on PMI in isolation. A higher-PMI continuous process can be more sustainable than a lower-PMI batch process if it delivers a significantly higher productivity (g of drug substance per unit time), thereby reducing energy consumption per unit produced [2].
- Verify Process Scalability: Lab-scale batch processes often face scaling difficulties that hurt material efficiency at commercial scale. Consider switching to a continuous process, which demonstrated a more consistent blend and better content uniformity in direct compression studies, leading to less waste [62].
- Assess Equipment Suitability: Batch processing equipment may be simpler but can lead to more wear and tear from frequent start-stop cycles and less consistent flow, increasing material variability and waste [29] [62]. Continuous processing equipment, while more complex, allows for real-time monitoring and control, enabling immediate correction of deviations that cause waste [67] [28].
Unexplained Cost Variance (COGS)
Problem: The actual Cost of Goods Sold (COGS) significantly exceeds the planned budget.
Solution: Decompose the total variance into its core components—volume, mix, and rate—to identify the root cause [70].
- Isolate Volume Variance: Use the formula:
Volume Variance = (Plan Quantity – (Total Actual Quantity * Plan Mix)) * Plan Rate[70]. A significant variance here means the total quantity of units sold differed from the plan. - Isolate Mix Variance: Use the formula:
Mix Variance = (Actual Quantity * (Actual Mix – Plan Mix)) * Plan Rate[70]. A significant mix variance indicates that the proportion in which different products were sold changed from the plan. Selling more of a high-cost product than planned creates an unfavorable mix variance. - Isolate Rate Variance: Use the formula:
Rate Variance = (Planned Rate – Actual Rate) X (Actual Quantity)[70]. An unfavorable rate variance is driven by increased costs per unit, such as vendor price increases on raw materials, higher transportation fees, or warehousing costs.
Frequently Asked Questions (FAQs)
Q1: From a sustainability perspective, is a continuous process always superior to a batch process?
Not necessarily. Research shows that the PMI of continuous manufacturing processes for biologics can be comparable to that of batch processes [2]. The overall sustainability assessment must look beyond PMI to include energy consumption, which can be lower for continuous processes due to higher productivity over time. Furthermore, continuous production is often cited as contributing to sustainability goals through reduced waste per unit and lower energy consumption [67] [29].
Q2: How does production volume directly influence the break-even point in cost analysis?
Production volume is the primary driver of the break-even point. The break-even point is the specific sales volume required to cover all fixed and variable costs, resulting in zero profit or loss [71]. As production volume increases, each unit sold contributes to covering the fixed costs (which remain constant). Once enough units are sold to cover these fixed costs, subsequent sales directly contribute to profit. This relationship is formally analyzed using Cost-Volume-Profit (CVP) analysis [71].
Q3: What are the key quality control differences when scaling an experiment from batch to continuous production?
The fundamental difference lies in the approach: discrete verification in batch versus continuous monitoring in continuous processes.
- Batch: Quality control is typically performed at the end of each production run on the completed batch. This allows for adjustments before the next batch begins [67] [29].
- Continuous: Quality control is integrated and performed in real-time throughout the production process. Automated systems and sensors (e.g., for temperature, pressure) constantly monitor critical quality attributes, allowing for immediate detection and correction of issues [67] [28]. A study on direct compression found that continuous processing resulted in a significantly lower variability for tablet mass and tensile strength compared to batch processing [62].
Q4: Can batch and continuous processes be combined?
Yes, many manufacturers implement semi-continuous processes that combine both methods. In this model, certain unit operations (e.g., mixing) may be performed in batches, with the outputs then fed into a continuous sequence for subsequent steps (e.g., tableting). This approach can offer a compromise between the flexibility of batch processing and the efficiency of continuous processing [28].
Experimental Data & Protocols
Quantitative Comparison: Batch vs. Continuous
The following table summarizes key comparative data from manufacturing research, particularly in pharmaceutical applications.
| Factor | Batch Process | Continuous Process | Key Research Findings |
|---|---|---|---|
| Production Volume & Speed | Slower; limited by batch size and setup times [28]. | Higher speed and volume through 24/7 operation [67] [28]. | Continuous processes are designed for high-volume, constant output [28]. |
| Cost Structure | Lower equipment cost, but higher unit cost due to lower rates and downtime [28]. | Higher equipment investment, but lower unit cost due to efficiency and scale [28]. | COGS variance analysis is critical for tracking volume, mix, and rate impacts in both systems [70]. |
| Process Mass Intensity (PMI) | Can be optimized but may face scale-up challenges. | Can be comparable to batch; overall sustainability may be better due to higher productivity [2]. | For biologics, continuous and batch PMI can be similar; energy/throughput is a key differentiator [2]. |
| Process Flexibility | High; easier to change products and formulations between batches [67] [28]. | Low; equipment is specialized for a specific product and changeovers are complex [28]. | Ideal for small runs and product testing [67]. |
| Quality Control (QC) | Discrete QC at the end of each batch step [67] [29]. | Real-time, continuous performance monitoring with sensors [67] [28]. | Continuous direct compression showed lower variability in tablet weight and tensile strength [62]. |
| Process Variability | Less consistent flow can result in higher in-process and quality variability [62]. | Better controlled flow leads to more consistent output and lower quality variability [62]. | In direct compression, batch processes showed "significantly higher variability" for key quality responses [62]. |
Experimental Protocol: Direct Compression Comparison
This protocol is adapted from a published study comparing batch and continuous direct compression for tablet manufacturing [62].
1. Objective: To directly compare the processability and final tablet quality (e.g., content uniformity, tensile strength, weight) of formulations produced via batch and continuous direct compression using a similar tablet press setup.
2. Materials:
- API: Paracetamol Powder (or relevant model drug).
- Fillers/Excipients: A selection of lactose types (e.g., Spray dried, Anhydrous, Granulated), Microcrystalline Cellulose (e.g., PH102).
- Other: Superdisintegrant (e.g., Primojel), Lubricant (e.g., Magnesium Stearate).
3. Equipment:
- Blender: For batch blend preparation (e.g., V-blender).
- Feed System: Hopper with rotating valve for batch processing; Continuous feeders for continuous line.
- Tablet Press: A modular rotary tablet press (e.g., GEA MODUL S) configured as similarly as possible for both batch and continuous runs.
- Analytical Tools: HPLC for content uniformity, hardness tester, balance.
4. Methodology:
- Formulation: Prepare low-dosed (e.g., 1% w/w) and high-dosed (e.g., 40% w/w) formulations.
- Blend Preparation (Batch): Weigh all components for a single batch. Blend in a V-blender. Transfer final blend to the batch process hopper.
- Blend Preparation (Continuous): This is an integrated process. Individual components are continuously fed via loss-in-weight feeders into a continuous blender, which discharges directly into the tablet press.
- Tableting: Run both processes at the same tablet press speed and forced feeder settings. Collect tablet samples at regular intervals throughout the run for analysis.
- Data Analysis: Use multivariate data analysis (e.g., Partial Least Squares regression) to correlate material properties, process parameters, and final tablet quality. Key is to analyze variance within the run (e.g., σMass for weight, σTS for tensile strength).
Process Visualization & Workflows
Batch vs. Continuous Production Workflow
Diagram 1: A comparison of discrete batch and uninterrupted continuous process workflows.
COGS Variance Analysis Decision Tree
Diagram 2: A troubleshooting tree for decomposing COGS variance into core components [70].
The Scientist's Toolkit: Key Research Reagents & Materials
The following table lists critical materials and their functions in comparative studies of batch and continuous manufacturing processes, particularly in solid dosage form development.
| Material / Solution | Function in Research Experiments | Relevance to Batch vs. Continuous |
|---|---|---|
| Spray-Dried Lactose (e.g., SuperTab 11SD) | Filler/Diluent; improves flowability and compressibility in direct compression formulations [62]. | Different lactose grades are tested in both processes to assess impact on flow and tablet quality variability [62]. |
| Microcrystalline Cellulose (e.g., Pharmacel 102) | Filler/Binder; provides high compactability and dry-binding properties [62]. | Often used in filler blends; its performance is compared across processes to evaluate content uniformity [62]. |
| Model API (e.g., Paracetamol Powder) | Active Pharmaceutical Ingredient used as a benchmark compound [62]. | Allows for a controlled comparison of content uniformity and process efficiency between batch and continuous lines [62]. |
| Sodium Starch Glycolate (e.g., Primojel) | Superdisintegrant; promotes tablet breakup in the gastrointestinal tract [62]. | A standard formulation component to ensure final product functionality is maintained during process comparison. |
| Magnesium Stearate | Lubricant; reduces friction during ejection from the tablet press die [62]. | Critical in both processes; over-lubrication can be a risk in continuous blending, requiring careful control of feeding rate [62]. |
For researchers and scientists in drug development, the Process Mass Intensity (PMI) metric has long been a standard for evaluating process efficiency. However, emerging research indicates that PMI alone provides an incomplete picture of environmental sustainability, particularly when comparing batch and continuous manufacturing processes for biologics. This technical resource center provides troubleshooting guidance and methodologies for comprehensively assessing environmental impact beyond PMI.
Key Metric Comparisons: PMI and Beyond
The following table summarizes critical environmental metrics that should be considered alongside PMI for a complete sustainability assessment:
| Metric | Definition | Batch Process Typical Values | Continuous Process Typical Values | Limitations |
|---|---|---|---|---|
| Process Mass Intensity (PMI) | Total mass input (kg) required to produce 1 kg of output [72] | ~7,700 kg/kg for mAbs [72] | Comparable to batch for mAbs [2] [26] | Does not account for energy, water for cleaning, or facility footprint [2] [72] |
| Cumulative Energy Demand (CED) | Total energy consumption across all process phases [72] | Higher per unit DS due to lower productivity and CIP/SIP operations [2] [72] | Potentially lower per unit DS due to higher productivity [2] [26] | Requires detailed tracking of all energy inputs |
| Global Warming Potential (GWP) | Greenhouse gas emissions expressed as CO₂ equivalent [72] | CIP/SIP can contribute to ~40% of GWP [72] | Generally lower due to reduced cleaning and smaller footprint [72] | Does not capture water or material consumption impacts |
| Water Consumption | Total water used in process, including buffers and cleaning [72] | 1.5+ million gallons/year for 20,000L facility; chromatography steps consume ~62% [72] | Significantly reduced; up to 95% reduction in buffer volumes in some unit operations [72] | PMI calculation often underestimates full water footprint |
Troubleshooting Common Experimental Challenges
Q: My data shows a continuous process with a higher PMI than batch, but literature suggests it should be more sustainable. What factors might explain this discrepancy?
A: PMI does not account for energy consumption, which can be a key driver of overall sustainability [2]. A continuous process with higher productivity (g of drug substance per unit time) can have lower overall energy consumption per unit produced, making it more environmentally sustainable despite a higher PMI [2] [26]. Focus on these additional metrics:
- Productivity rates: Calculate output per unit time rather than per batch [2]
- Energy monitoring: Install sub-meters to track energy use by major equipment
- Lifecycle stages: Evaluate supply chain, use phase, and end-of-life impacts [72]
FAQ: Addressing Water Consumption Inefficiencies
Q: My continuous bioreactor process shows unexpectedly high water consumption. Where should I focus optimization efforts?
A: High water usage in continuous processes often stems from these common issues:
- Buffer preparation: Chromatography steps consume 62% of water in biologics processes [72]
- Media optimization: Perfusion bioreactors may need more media, but this can be offset with N-1 perfusion intensification [72]
- Final formulation: The last step in continuous mAb processes may have higher buffer consumption than batch diafiltration [72]
Mitigation strategies:
- Implement flow-through chromatography to reduce buffer volumes [72]
- Switch to membrane chromatography instead of resin columns [73]
- Optimize media for perfusion processes to reduce consumption [72]
Experimental Protocols for Comprehensive Environmental Assessment
Protocol 1: Comparative Life Cycle Assessment (LCA) for Biologics Processes
Objective: Conduct a standardized LCA comparing batch and continuous manufacturing processes to evaluate overall environmental impact beyond PMI.
Methodology:
- System Boundaries: Define boundaries across three phases [72]:
- Supply chain phase (raw material production and transportation)
- Use phase (actual production, including CIP/SIP operations)
- End-of-life phase (disposal, reuse, and recycling)
Data Collection:
- Track all material inputs with precise weighing systems
- Install energy meters on all major equipment (bioreactors, chromatography skids, purification systems)
- Monitor water sources (municipal, WFI) with flow meters
- Document waste streams (biological, chemical, recyclables)
Impact Categories [72]:
- Global Warming Potential (GWP)
- Cumulative Energy Demand (CED)
- Water Scarcity Impact
- Resource Depletion
Calculation:
- Normalize all inputs per kg of drug substance (DS) produced
- Compare batch vs. continuous using statistical analysis (t-test, p<0.05)
- Perform sensitivity analysis on key parameters
Protocol 2: Energy Consumption Analysis During Process Intensification
Objective: Quantify energy savings from continuous process intensification despite comparable PMI values.
Methodology:
- Experimental Setup:
- Configure identical bioreactor scales for batch and continuous processes
- Operate continuous process at multifold higher productivity rates
- Maintain equivalent quality specifications for both processes
Data Collection:
- Record energy consumption (kWh) using calibrated power meters
- Track active production time vs. downtime for each method
- Document CIP/SIP cycles for batch processes [72]
- Monitor environmental controls (HVAC) energy usage
Calculation:
- Compute energy consumption per gram of drug substance (kWh/g DS)
- Compare despite PMI equivalence using the formula:
- Factor in reduced cleaning requirements for continuous processes [72]
The Scientist's Toolkit: Essential Research Reagents and Solutions
| Item | Function | Application Notes |
|---|---|---|
| Process Analytical Technology (PAT) | Real-time monitoring of critical quality attributes [74] | Essential for continuous process control; requires spectroscopic tools and data modeling expertise |
| Single-Use Technologies (SUT) | Disposable bioreactors, filters, and connectors [72] | Eliminates CIP/SIP requirements, reducing water and energy consumption; particularly beneficial for continuous processes |
| Membrane Chromatography | Purification technology replacing resin columns [72] | Reduces buffer consumption by up to 90% compared to batch operations |
| N-1 Perfusion Systems | High-density seed train intensification [72] | Increases inoculum density, shortening production reactor time and saving media |
| Flow-Through Chromatography | Continuous purification method [72] | Eliminates washing and elution steps, significantly reducing buffer utilization |
Advanced Technical Guidance
Implementing Effective Process Analytical Technology
Continuous manufacturing requires sophisticated PAT for real-time quality verification [74]. Common challenges and solutions include:
- Content uniformity monitoring: Particularly difficult for low API concentrations; requires high time-resolution measurements [74]
- Dosing verification: Loss-in-weight feeders run blind during refill; implement redundant measurement systems like spectrometers [74]
- Data synchronization: Analytical responses must correlate with equipment parameters, correcting for sampling lags [74]
Strategic Decision Framework for Process Selection
When evaluating batch versus continuous processes, consider this analytical framework:
Product-Specific Factors:
Environmental Impact Considerations:
Hybrid Approaches:
- Many facilities successfully combine continuous base production with batch finishing steps [75]
- This approach balances efficiency with flexibility for product variations
Moving beyond PMI as a sole metric requires researchers to adopt comprehensive assessment methodologies that incorporate energy consumption, water usage, and overall environmental impact. The protocols and troubleshooting guides provided here enable scientists to make more informed decisions when comparing batch and continuous processes, ultimately leading to more sustainable biopharmaceutical manufacturing.
Assessing Operational Flexibility and Robustness to Supply Chain Shocks
Frequently Asked Questions (FAQs)
Q1: How do batch and continuous processes differ in their ability to respond to sudden disruptions in the supply of raw materials? Batch processes offer greater short-term flexibility to absorb supply shocks. You can adjust recipes, substitute approved alternative raw materials, or temporarily reduce batch sizes without halting an entire production line [4] [44]. Continuous processes, while efficient, are more vulnerable. They require a constant, reliable flow of raw materials; any interruption can force a full shutdown and restart, which is time-consuming and costly [27] [44].
Q2: Which process offers greater flexibility for producing multiple products or product variants? Batch processing is significantly more flexible for multi-product facilities. Equipment can be cleaned and reconfigured between batches to produce different formulations, making it ideal for personalized medicines or a diverse portfolio of specialty chemicals [67] [4] [28]. Continuous processes are designed for long-term production of a single product and lack this agility [4].
Q3: How does each process handle scaling production up or down to meet volatile demand? Batch processes allow for "scale-out" strategies. You can meet increased demand by adding another batch reactor or duplicating production lines, which is often more capital-efficient [44]. Continuous processes are designed for a specific capacity. Scaling up typically requires extending run times (which has limits due to equipment wear) or a major capital project to redesign the line, making it less responsive to sudden demand changes [27] [44].
Q4: From a quality control perspective, which process is more robust against producing large quantities of non-conforming product? Batch processing contains quality events within a single, discrete batch. If a deviation is detected, only that specific batch must be rejected or reworked [29] [6]. In continuous processing, a process deviation can affect all product manufactured from the time the error occurred until it is detected and corrected by the control system, potentially leading to a larger volume of waste [6].
Troubleshooting Guides
Issue: Inconsistent Product Quality Between Batches
Problem: Final product quality varies significantly from one batch to another.
- Potential Cause 1: Inconsistent raw material properties or operator-dependent manual procedures [6].
- Solution: Implement stricter raw material qualification and develop detailed Standard Operating Procedures (SOPs) to minimize human variability.
- Potential Cause 2: Equipment not adequately cleaned or calibrated between batches.
- Solution: Enforce rigorous cleaning validation protocols and a preventive maintenance schedule.
Issue: Unplanned Downtime in a Continuous Process Line
Problem: A disruption in raw material supply or a minor equipment failure halts the entire continuous line.
- Potential Cause 1: Lack of buffer capacity or surge bins between unit operations.
- Solution: Redesign the line to include small buffer tanks to temporarily decouple unit operations, allowing one section to be serviced without stopping the entire line.
- Potential Cause 2: Inadequate real-time monitoring to predict equipment failures.
Issue: Inability to Scale a Batch Process Efficiently
Problem: A successful laboratory-scale batch process cannot be reproduced at a commercial scale.
- Potential Cause: Scale-up is not a linear process; factors like mixing efficiency and heat transfer change with volume.
- Solution: Employ Quality by Design (QbD) principles early in development to understand the critical process parameters (CPPs) that impact scale-up. Consider transitioning to a continuous process, which scales more linearly by simply extending runtime [27].
Quantitative Data Comparison
The following tables summarize key operational and performance indicators for batch and continuous processes, providing a data-driven basis for assessing robustness.
Table 1: Key Performance Indicators for Batch and Continuous Processes
| Performance Indicator | Batch Process | Continuous Process | Source |
|---|---|---|---|
| Production Volume | Suitable for small to medium volumes [4] | Ideal for high-volume, consistent output [4] | [4] |
| Relative Cost Structure | Lower initial setup cost, but higher unit costs [28] | High initial investment, but lower unit costs at scale [28] | [28] |
| Process Flexibility | High; easy to reconfigure for different products [67] [4] | Low; designed for a specific product [4] | [67] [4] |
| Quality Control Approach | Testing at the end of a batch [28] | Real-time monitoring with PAT tools [67] [6] | [67] [28] [6] |
| Impact of a Quality Failure | One single batch is affected [29] [6] | All product from the deviation period is affected [6] | [29] [6] |
Table 2: Operational Resilience and Supply Chain Indicators
| Resilience Indicator | Batch Process | Continuous Process |
|---|---|---|
| Response to Raw Material Shortage | Can adjust batch size or formulation; more adaptable to intermittent supply [44] | Highly vulnerable; requires constant material flow, shutdowns are costly [44] |
| Speed to Increase Output | Slow; requires "scaling out" with new equipment [44] | Fast for small increases (extend runtime); slow for large increases (requires new line) [27] |
| Sustainability & Waste | Higher energy use from frequent start/stops; potentially more waste per unit [76] | Reduced energy and material waste per unit; supports sustainability goals [67] [29] |
| Typical Industries | Pharmaceuticals, Specialty Chemicals, Nanomaterials, Food & Beverage [67] [44] | Commodity Chemicals, Metal Fabrication, High-Volume Food Production [67] [44] |
Experimental Protocols
Protocol: Direct Comparison of Batch and Continuous Direct Compression for Tablet Manufacturing
Objective: To directly compare the processability and final tablet quality (e.g., content uniformity, tensile strength) of a formulation when processed via batch and continuous direct compression methods using similar equipment.
Background: This protocol is based on a study that filled a key research gap by using a similar tablet press configuration for both methods, allowing for a more accurate comparison [62].
Materials:
- API: Paracetamol Powder (or model API of choice) [62]
- Fillers: e.g., Spray-dried lactose (SuperTab 11SD), Anhydrous lactose (SuperTab 22AN), Microcrystalline cellulose (Pharmacel 102) [62]
- Disintegrant: Sodium starch glycolate (Primojel) [62]
- Lubricant: Magnesium Stearate [62]
Methodology:
- Formulation: Prepare two sets of blends for each process:
- Low-dose: 1% w/w API, 94% w/w filler, 4% w/w disintegrant, 1% w/w lubricant.
- High-dose: 40% w/w API, 55% w/w filler, 4% w/w disintegrant, 1% w/w lubricant [62].
- Batch Direct Compression:
- Pre-blend all powders in a suitable batch blender (e.g., a V-blender) for a fixed time.
- Transfer the final blend to a hopper feeding a stand-alone rotary tablet press.
- Compress tablets at a defined speed and pressure [62].
- Continuous Direct Compression (CDC):
- Use an integrated CDC line with individual loss-in-weight feeders for the API, excipients, and lubricant.
- Feed materials continuously into a continuous blender (e.g., a twin-screw blender).
- Direct the resulting blend to an integrated rotary tablet press.
- Compress tablets using the same tooling and similar press settings as the batch process [62].
- Analysis:
- Processability: Monitor feeder accuracy, blend homogeneity (using PAT probes in continuous line), and tablet press variability (e.g., mass control).
- Tablet Quality: Sample tablets at regular intervals and analyze for weight uniformity, API content uniformity, tensile strength, and dissolution [62].
Process Flow Visualization
Batch vs. Continuous Process Flow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Solid Dosage Form Manufacturing Research
| Material / Reagent | Function in Research & Development | Key Considerations for Process Selection |
|---|---|---|
| Spray-Dried Lactose (e.g., SuperTab 11SD) | A common filler (diluent) in tablet formulations. | Excellent flowability is critical for both batch and continuous processes, but is especially important for consistent feeding in continuous direct compression (CDC) [62]. |
| Microcrystalline Cellulose (e.g., Pharmacel 102) | A versatile filler and binder that also promotes disintegration. | Known for good compressibility and consistent performance. Its properties are less dependent on process type, making it a robust choice for process comparison studies [62]. |
| Anhydrous Lactose (e.g., SuperTab 22AN) | A direct compression filler. | Material properties related to flow, compressibility, and permeability are crucial differentiators between batch and continuous processes in research settings [62]. |
| Sodium Starch Glycolate (e.g., Primojel) | A super-disintegrant to promote tablet breakdown in the gastrointestinal tract. | Its function is largely process-agnostic, but its concentration and distribution (content uniformity) are key quality attributes to measure when comparing batch and continuous blending efficacy [62]. |
| Magnesium Stearate | A lubricant to prevent sticking to tooling during compression. | Lubrication efficiency and potential over-lubrication can be process-dependent. Mixing time and shear in a continuous blender vs. a batch blender can significantly impact tablet tensile strength and dissolution [62]. |
The following table synthesizes key quantitative findings from comparative research on batch and continuous manufacturing processes for biologics, focusing on Process Mass Intensity (PMI) and related operational factors.
Table 1: Comparative Performance of Batch and Continuous Biologics Manufacturing Processes [2] [26]
| Performance Metric | Batch Process | Continuous Process | Context & Implications |
|---|---|---|---|
| Process Mass Intensity (PMI) | Comparable to continuous processes [2] [26] | Comparable to batch processes [2] [26] | PMI alone is an insufficient sustainability metric; it does not account for energy consumption, a key driver of environmental impact [2]. |
| Productivity (g of Drug Substance per unit time) | Baseline | Multifold higher [2] | Higher productivity can render a continuous process with a higher PMI more environmentally sustainable than a batch process due to lower overall energy consumption per unit of DS produced [2]. |
| Impact of Process Intensification | Not specified | Drives significant improvement in sustainability [2] | Intensification strategies can substantially enhance the material usage efficiency and overall sustainability profile of continuous processes [2]. |
Experimental Protocols for Process Comparison
Protocol: Calculating and Comparing Process Mass Intensity (PMI)
Objective: To quantify and compare the material usage efficiency of batch and continuous manufacturing processes for monoclonal antibodies (mAbs) [2] [26].
Methodology:
- System Boundary Definition: Define the scope of the manufacturing process to be analyzed, typically from the initial cell culture steps to the final purified drug substance.
- Total Mass Input Calculation: Sum the mass of all materials fed into the process, including water, cell culture media, buffers, and purification resins.
- Drug Substance Mass Determination: Measure the total mass of the final output, which is the purified drug substance (DS).
- PMI Calculation: Calculate the Process Mass Intensity using the formula: PMI = Total Mass of Inputs (kg) / Mass of Drug Substance (kg) [2] [26].
- Comparative Analysis: Perform this calculation for both a standard batch process and a continuous process. Conduct a sensitivity analysis to understand how different process parameters (e.g., perfusion rate, harvest cell density) impact the PMI of the continuous system [2].
Protocol: Assessing Sustainability Beyond PMI
Objective: To evaluate the environmental sustainability of a manufacturing process by integrating energy consumption data with PMI [2].
Methodology:
- Energy Consumption Profiling: Measure the total energy consumed (e.g., in kWh) by the manufacturing process over a defined operational period for both batch and continuous systems. This includes energy for bioreactor agitation, temperature control, purification, and water purification.
- Productivity-Linked Functional Unit: Define the functional unit for comparison as the energy consumption per gram of drug substance produced.
- Integrated Analysis: Compare the batch and continuous processes using this functional unit. A continuous process with a higher PMI but significantly higher productivity may demonstrate a lower environmental impact per gram of DS when energy consumption is factored in [2].
Troubleshooting Guides & FAQs
Frequently Asked Questions
Q1: Our calculated PMI for a new continuous process is higher than our established batch process. Does this mean the continuous process is less sustainable? A: Not necessarily. PMI measures material efficiency but excludes energy consumption, a critical sustainability factor. A continuous process often has multifold higher productivity (grams of drug per unit time). You must calculate the energy consumption per gram of drug substance. A continuous process can be more sustainable overall if its higher productivity leads to lower energy use per unit of product, even with a higher PMI [2].
Q2: When investigating process performance issues, where should we start? A: Begin by clarifying the process's fundamental purpose using a Value Proposition Statement. This ensures your investigation is aligned with what the process is meant to deliver. Next, use tools like the Kano Model to ensure you understand critical customer (or patient) needs. Finally, establish Operational Definitions for all key metrics (e.g., "on-time delivery") to ensure all stakeholders are measuring and interpreting data consistently, preventing misdiagnosis of problems [77].
Q3: How can we balance quality with efficiency when optimizing a process? A: Do not compromise quality for efficiency. Methodologies like Six Sigma DMAIC (Define, Measure, Analyze, Improve, Control) can be used to embed quality directly into process optimization projects. This structured approach ensures that improvements to speed or cost do not increase defects or variations in the final product [78].
Process Selection Troubleshooting Guide
Problem: High Material Consumption in Development
- Check 1: Calculate the PMI for your process and benchmark it against industry standards for both batch and continuous modes [2] [26].
- Check 2: Perform a sensitivity analysis on your process parameters. For a continuous process, factors like perfusion rate can significantly impact material usage efficiency [2].
- Solution: Explore process intensification strategies. Research indicates that intensification is a key driver for improving the sustainability of biologics manufacturing, potentially offsetting high material use through greater output [2].
Problem: Uncertainty in Long-Term Process Sustainability
- Check 1: Confirm you are looking beyond PMI. A myopic focus on mass intensity can lead to suboptimal environmental decisions [2].
- Check 2: Integrate energy consumption data into your model using the experimental protocol in Section 2.2.
- Solution: Advocate for the development and use of comprehensive sustainability metrics and models that account for energy, water, and other environmental factors alongside traditional efficiency metrics [2].
Research Reagent Solutions & Essential Materials
Table 2: Key Research Reagents and Materials for Process Development [2] [26]
| Reagent/Material | Function in Process Development |
|---|---|
| Cell Culture Media | Supports the growth and viability of production cell lines (e.g., CHO cells) in the bioreactor stage. Its efficient use is a major factor in PMI [2] [26]. |
| Purification Resins & Chromatography Buffers | Critical for downstream purification of the target biologic (e.g., mAb) from process impurities. The quantity and reuse cycles of resins directly impact material consumption and PMI [2]. |
| Monoclonal Antibodies (mAbs) | The primary therapeutic product model used in the cited comparative studies. Serves as the "output" in the PMI calculation formula [2] [26]. |
| Drug Substance (DS) | The final, purified output of the manufacturing process. The mass of DS is the denominator in the PMI calculation and the basis for productivity comparisons [2]. |
Process Analysis Workflow Diagram
Conclusion
The comparison between batch and continuous manufacturing reveals a nuanced landscape where PMI is a crucial, but not solitary, metric. While batch processes may show lower PMI in specific, smaller-scale scenarios, continuous manufacturing presents a compelling case for large-volume production with its potential for significant operational cost savings, enhanced quality control, and superior scalability. Future success hinges on developing comprehensive sustainability models that integrate PMI with energy and cost metrics, fostering regulatory alignment, and advancing process intensification technologies. For biomedical research, embracing continuous processing can accelerate drug development timelines, reduce the environmental footprint of clinical manufacturing, and enhance the agility of supplying therapies for clinical trials and the market.
References
- 1. Process Mass Intensity (PMI): A Holistic Analysis of Current ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11002941/]
- 2. Comparison of process mass intensity (PMI) of continuous ... [https://www.sciencedirect.com/science/article/pii/S1871678422000619]
- 3. Batch Manufacturing: Examples & Tips For Efficiency [https://www.fishbowlinventory.com/blog/batch-manufacturing]
- 4. Batch Process vs Continuous Process: Key Differences ... [https://snicsolutions.com/blog/batch-process-vs-continuous-process]
- 5. What is Batch Manufacturing? Processes, Benefits, and ... [https://snicsolutions.com/blog/what-is-batch-manufacturing]
- 6. Continuous vs. Batch Manufacturing: What's the Difference? [https://www.news-medical.net/life-sciences/Pharmaceutical-Continuous-Manufacturing-vs-Batch-Manufacturing-Whats-the-Difference.aspx]
- 7. Batch Production: Examples, Advantages and Disadvantages [https://www.projectmanager.com/blog/batch-production]
- 8. Running and Troubleshooting Batch Processing [https://docs.oracle.com/en/industries/energy-water/shared/22b/cs-live-ops-guide/CS_LIVE_OPS_22B/Cloud_Live_Operate_Troubleshooting.7.3.html]
- 9. The Top 20 Problems with Batch Processing (and How to ... [https://www.kai-waehner.de/blog/2025/04/01/the-top-20-problems-with-batch-processing-and-how-to-fix-them-with-data-streaming/]
- 10. 5 Key Principles of Continuous Flow in Lean Manufacturing [https://www.sixsigmaonline.org/continuous-flow-lean/]
- 11. Exploring Continuous Pharmaceutical Manufacturing vs. ... [https://clarkstonconsulting.com/insights/continuous-pharmaceutical-manufacturing/]
- 12. Continuous Flow Manufacturing: A Guide to Streamlined ... [https://www.6sigma.us/manufacturing/continuous-flow-manufacturing-cfm/]
- 13. Troubleshooting in pharmaceutical manufacturing processes [https://www.hwi-group.de/en/blog/details/troubleshooting-in-pharmaceutical-manufacturing-processes-root-cause-analysis-of-quality-defects]
- 14. Preventing and Troubleshooting Manufacturing Deviations [https://www.pharmtech.com/view/preventing-and-troubleshooting-manufacturing-deviations]
- 15. The Five Principles of Lean [https://theleanway.net/The-Five-Principles-of-Lean]
- 16. Sustainability case studies on the use of continuous ... [https://www.sciencedirect.com/science/article/pii/S2666086522000212]
- 17. Sustainability in Projects [https://www.pmi.org/learning/sustainability]
- 18. Data Visualizations, Charts, and Graphs - Digital Accessibility [https://accessibility.huit.harvard.edu/data-viz-charts-graphs]
- 19. Data Visualization For Business Analysts [https://www.pmi.org/learning/library/data-visualization-business-analysts-10198]
- 20. PharmaPy: An object-oriented tool for the development of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10793241/]
- 21. Welcome to PharmaPy's documentation! — PharmaPy 2023 ... [https://pharmapy.readthedocs.io/]
- 22. Batch vs. Continuous Bulk Processing: Pros and Cons [https://www.bpsvibes.com/blog/batch-vs-continuous-bulk-processing-pros-and-cons]
- 23. What are Critical Quality Attributes (CQAs)? [https://www.hamiltoncompany.com/knowledge-base/article/what-are-critical-quality-attributes-cqas?srsltid=AfmBOorQztkQf_JXc_er9IJudv0zYFOGXzH5FKCVpA4Wf7jAXM-InvcH]
- 24. What are CQAs?: Understanding the FDA's Critical Quality ... [https://mkainsights.com/what-are-cqas-understanding-the-fdas-critical-quality-attributes/]
- 25. Defining Critical Quality Attributes in the Pharmaceutical ... [https://www.gxp-cc.com/insights/blog/defining-critical-quality-attributes-in-the-pharmaceutical-manufacturing-process/]
- 26. Comparison of process mass intensity (PMI) of continuous ... [https://pubmed.ncbi.nlm.nih.gov/36368463/]
- 27. Continuous manufacturing versus batch ... [https://gabi-journal.net/continuous-manufacturing-versus-batch-manufacturing-benefits-opportunities-and-challenges-for-manufacturers-and-regulators.html]
- 28. Continuous vs. Batch Process: What Are the Differences? [https://katanamrp.com/blog/continuous-vs-batch-process/]
- 29. Batch Processing vs. Continuous Processing [https://www.getmaintainx.com/learning-center/batch-processing-vs-continuous-processing]
- 30. Integration of techno-economic analysis and life cycle ... [https://www.sciencedirect.com/science/article/abs/pii/S0959652621024641]
- 31. Sustainability and Techno-Economic Assessment of Batch ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11864096/]
- 32. Continuous pharmaceutical manufacturing and its ... [https://link.springer.com/article/10.1007/s42452-025-07712-9]
- 33. An open-source platform for surrogate techno economic ... [https://www.sciencedirect.com/science/article/pii/S0098135425002248]
- 34. Comparative analysis of tablet dissolution behavior: Batch ... [https://www.sciencedirect.com/science/article/abs/pii/S0378517325003357]
- 35. Stochastic Technoeconomic Analysis and Optimization of ... [https://www.sciencedirect.com/science/article/abs/pii/B9780443288241502337]
- 36. Process modelling, design and technoeconomic evaluation ... [https://www.sciencedirect.com/science/article/abs/pii/S0098135418301984]
- 37. Effect of Choice of Solvent on Crystallization Pathway ... [https://www.mdpi.com/2073-4352/10/12/1107]
- 38. Crystallization of Form II Paracetamol with the Assistance of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9147162/]
- 39. 3.6F: Troubleshooting [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_Lab_Techniques_(Nichols)/03%3A_Crystallization/3.06%3A_Step-by-Step_Procedures/3.6F%3A_Troubleshooting]
- 40. Troubleshooting Tips for Crystallization Processes [https://www.linkedin.com/advice/0/how-can-you-troubleshoot-crystallization-process-skztf]
- 41. Cooling crystallization monitoring and control in API ... [https://www.vaisala.com/en/blog/2025-09/cooling-crystallization-monitoring-and-control-api-production-processes-ri-measurements]
- 42. (655c) Development of a Solubility Measurement Workflow ... [https://proceedings.aiche.org/conferences/aiche-annual-meeting/2024/proceeding/paper/655c-development-solubility-measurement-workflow-accelerating-crystallization-process-development]
- 43. Continuous vs. Batch Manufacturing in Pharmaceuticals [https://www.pharmafocusamerica.com/manufacturing/continuous-batch-m00anufacturing]
- 44. Batch vs. Continuous Manufacturing [https://cerionnano.com/2023/11/batch-vs-continuous-manufacturing-is-the-nanomaterials-industry-lagging-or-leading/]
- 45. Pharmaceutical Process scale-up from a Lab mill to ... [https://www.quadro-mpt.com/news-and-events/process-scale-up-and-technology-transfer]
- 46. How to Scale Up from Lab to Production: A Guide ... [https://www.maxwell-machine.com/how-to-scale-up-from-lab-to-production]
- 47. Validating a Pharmaceutical Research and Development ... [https://www.pmi.org/learning/library/validating-pharmaceutical-research-development-facility-1051]
- 48. Guide of Pilot Plant Scale-Up Techniques [https://adesisinc.com/a-comprehensive-guide-to-pilot-plant-scale-up-techniques/]
- 49. Continuous vs. Batch Process: What are the Differences? [https://ajawmrp.com/continuous-vs-batch-process-what-are-the-differences/]
- 50. A Comparative Investment Analysis of Batch Versus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8802271/]
- 51. Complexity of Project Management in the Pharmaceutical ... [https://www.pmi.org/learning/library/project-management-complexity-pharmaceutical-industry-1487]
- 52. Process Mass Intensity Calculator [https://acsgcipr.org/tools/process-mass-intensity/]
- 53. Batch vs. Continuous Processes [https://appliedchemical.com/services/research-development/pilot-plant/batch-vs-continuous/]
- 54. Evolving the Green Chemist's Toolbox for APIs Manufacturing [https://www.seqens.com/knowledge-center/evolving-the-green-chemists-toolbox-for-apis-manufacturing/]
- 55. Chemical Engineering & Process Intensification [https://www.chemcopilot.com/blog/chemical-engineering-process-intensification-rethinking-scale-sustainability-and-speed]
- 56. Process intensification: A game changer for the pharma ... [https://www.openaccessgovernment.org/article/process-intensification-a-game-changer-for-the-pharma-market/188486/]
- 57. Process intensification control: Advancing efficiency and ... [https://www.sciencedirect.com/science/article/abs/pii/S0255270125002375]
- 58. Modular Control of an Integrated Continuous Synthesis ... [https://www.sciencedirect.com/science/article/abs/pii/S0263876225005659]
- 59. Positive Material Identification (PMI) Guide for Alloy ... [https://www.lgcstandards.com/US/en/Resources/Articles/positive-material-identification]
- 60. Barriers to Organizational Project Management [https://www.pmi.org/learning/library/barriers-organizational-project-management-7405]
- 61. Regulatory Compliance in Pharma Manufacturing [https://www.pharmanow.live/pharma-manufacturing/challenges-strategies-pharma-manufacturing]
- 62. Batch vs. continuous direct compression – a comparison of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10792456/]
- 63. Pharma 4.0 and Validation 4.0 Strategies for Compliance [https://pscsoftware.com/pharma-4-validation-4-strategies-compliance/]
- 64. Real-Time Monitoring: Why Have It and How to Get It [https://www.valgenesis.com/blog/real-time-process-monitoring-why-you-need-it-and-how-to-get-it]
- 65. A Guide to Pharma 4.0: Embracing Transformation in the ... [https://learn.g2.com/pharma-4.0]
- 66. Implementing Pharma 4.0 Solutions for Fill-Finish [https://www.ast-inc.com/blogs/5-questions-on-implementing-pharma-four-point-zero-solutions-for-aseptic-fill-finish/]
- 67. Batch vs Continuous Manufacturing Production | ATS [https://www.advancedtech.com/blog/batch-vs-continuous-manufacturing/]
- 68. Designing for Pharma 4.0 [https://www.pmgroup-global.com/news-insights/designing-for-pharma-4-0/]
- 69. Continuous vs. Batch Processing for Biopharma ... [https://hp-ne.com/blog/continuous-vs-batch-processing-for-biopharma-manufacturing/]
- 70. Manufacturing COGS Variance: Volume, Mix, Rate [https://8020consulting.com/blog/manufacturing-cogs-variance-volume-mix-rate]
- 71. Cost Volume Profit Analysis in Manufacturing Businesses [https://accounovation.com/blogs/cost-volume-profit-analysis-in-manufacturing-businesses]
- 72. What's The Environmental Impact Of Biopharma ... [https://www.bioprocessonline.com/doc/what-s-the-environmental-impact-of-biopharma-continuous-manufacturing-part-i-0001]
- 73. The difference between batch, fed-batch, and continuous ... [https://infors-ht.com/en/blog/the-difference-between-batch-fed-batch-and-continuous-processes]
- 74. Continuous Versus Batch: A Pharmaceutical Manufacturing ... [https://www.tabletscapsules.com/3641-Technical-Articles/581452-Continuous-Versus-Batch-Pharmaceutical-Manufacturing-Q-A/]
- 75. Batch Process vs Continuous Process for Food Manufacturers [https://tulip.co/blog/batch-process-vs-continuous-process/]
- 76. How Batch Processing Differs From Continuous Flow ... [https://www.generalkinematics.com/blog/batch-processing-vs-continuous-flow/]
- 77. Which Tools Work Best for Understanding Process Problems? [https://pmi.co.uk/knowledge-hub/which-tools-work-best-for-understanding-process-problems/]
- 78. Business process optimization [https://www.pmi.org/learning/library/optimization-project-management-six-sigma-8010]